 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christina Vasilopoulou | + 3336 word(s) | 3336 | 2020-11-30 09:11:01 | | | |
| 2 | Christina Vasilopoulou | Meta information modification | 3336 | 2020-12-09 17:42:18 | | | | |
| 3 | Rita Xu | Meta information modification | 3336 | 2020-12-10 04:39:40 | | |
Video Upload Options
Amyotrophic Lateral Sclerosis (ALS) is the most common late-onset motor neuron disorder, but our current knowledge of the molecular mechanisms and pathways underlying this disease remains elusive. Genome-Wide Association Studies (GWAS) aim to identify Single Nucleotide Polymorphisms (SNPs) and other types of genetic variation that are more frequent in patients than in people without the disease, using a variety of statistical tests. Despite the rapid recent technological advances and great efforts in the GWAS field that have led to the genomic profiling of large ALS cohorts, the identified associations have been able to explain only a very small fraction of the ALS heritability and aetiology. Here, we outline ALS-Specific GWAS Challenges, explaining the limitations of traditional GWAS analyses, considering known features of the ALS genetic architecture and hypotheses about ALS pathology (e.g., multilocus interactions, rare variations with low effect size). Future advances in the genomic and machine learning fields may bring about a better understanding of ALS genetic architecture and enable improved personalized approaches to this and other devastating and complex diseases.
1. Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a progressively fatal, late-onset motor neuron disorder that is predominately characterised by the loss of upper and lower motor neurons. Progressive muscle atrophy in ALS patients leads to swallowing difficulties, paralysis and ultimately to death from neuromuscular respiratory failure [1][2][3]. ALS is the most common type of motor neuron disorder, and has peak onset at 54–67 years old, although it can affect individuals of any age [2][3][4][5]. Patients typically survive 2–5 years after the first symptoms occur, with 5–10% surviving more than 10 years [1][2][6]. A population-based study of estimated ALS incidence in 10 countries found that prevalence could increase more than 31% from 2015 to 2040 [4]. Thus, there is an increasing need to understand ALS pathology and the molecular pathways involved, towards prevention or successful therapeutic intervention.
There are two major classifications among ALS patients, based on family history: 5–10% of cases are genetically linked, and are classified as familial, having one or more relatives that suffer from ALS, while 90% are classified as sporadic, in which a familial history is not established, and where a genetic cause is usually not identified [7]. However, the distinction between the two categories is not always simple, with familial ALS-associated mutations also being present among sporadic ALS cases [3]. The extent and form of genetic contribution to sporadic ALS remains unclear, but genetic factors are considered to play an important role in the disease pathology [8]. Further investigation of the genetic architecture of both familial and sporadic cases is necessary.
1.1. Current Knowledge of Molecular Pathways Implicated by the Functions of Known ALS-Linked Genes
Our current knowledge of the aetiology and the genetic architecture of ALS is still elusive. Genetic mutations, environmental contributions, epigenetic changes and DNA damage are hypothesized as potential causal factors that ultimately lead to motor neuron death [9][10]. Variants in more than 30 genes are recognized as monogenic causes of ALS [9][11][12][13][14]. The most frequent monogenic cause in European populations is the intronic hexanucleotide GGGGCC (G4C2) repeat expansion (HRE) in the C9orf72 gene [15][16]. Other genes linked to ALS with high reproducibility include Cu/Zn superoxide dismutase 1 SOD1, fused in sarcoma FUS, and transactive response DNA-binding protein of 43 kD TARDBP/TDP-43 [9]. The discovery of risk gene mutations has helped to unravel the molecular mechanisms of ALS, and may lead ultimately to targeted therapy and stratified drug discovery [9][17][18][19].
Numerous studies have been published aimed at explaining motor neuron death, investigating the functional effects of specific mutations of known risk-associated genes such as C9orf72, FUS, SOD1 and TDP-43 [14][18]. Recent systematic reviews from our group have summarised the molecular pathways and biomarkers for which there are strong supporting evidence in ALS [9][17][18]. The molecular pathways affected in ALS can be grouped as follows (see [18] for detailed review):
-
Mitochondrial dysfunction as a direct or indirect consequence of ALS-associated gene mutations CHCHD10, FUS, SOD1, C9orf72 and TDP-43 can lead to an increase in oxidative stress, an increase in cytosolic calcium, ATP deficiency and/or stimulation of pro-apoptotic pathways [10][13][18][20][21].
-
Oxidative stress can also be derived from a stimulation of NADPH oxidase, as observed with ATXN2 mutations [22], or from deficiency in the elimination of Reactive Oxygen Species (ROS) as observed with some SOD1 mutations in familial cases [18][23][24]. It then may contribute to DNA damage. Interestingly, other mutations on the ALS-associated genes NEK1 [25], SETX [26] and C21orf2 [27] are suspected to alter the DNA repair machinery, leading to an accumulation of oxidative damage over time. Consequently, these events could ultimately lead to motor neuron death [25][28].
-
Disrupted axonal transport has been directly linked to a mutation in the C-terminal of the ALS-associated gene, KIF5A [11][29], and to mutations in genes encoding for neurofilaments (NEFH), microtubules and motor proteins (PFN1,TUBA4A, DCTN1) [18][30]. Consequently, organelle transport, protein degradation, and RNA transport are affected, disrupting cellular homeostasis. Similarly, axonal transport disruptions have been observed in fALS patients harboring mutations in non-cytoskeletal-related genes such as SOD1 [29].
-
Protein degradation is suspected to be a key pathway that is defective in ALS. This can be a direct consequence of mutations in ALS-associated genes involved in proteasome activity and the autophagy pathway, such as UBQLN2, VCP, SQSTM1/P62, OPTN, FIG4, Spg11, or TBK1 [31], and may lead to an accumulation of misfolded and non-functional proteins [18][32]. It can also be an indirect consequence of other mutations leading to the formation of protein aggregates such as SOD1, FUS, TDP43, C9orf72-derived DPR - aggregates that in turn impair the proteasome and autophagic degradation pathways [33], thus exacerbating the accumulation of misfolded proteins. Consequently, the blockade of autophagy pathways may affect vesicle secretion [34][35]. Interestingly, some ALS-associated genes are known to be directly or indirectly involved in exosome biogenesis such as CHMP2B [36] or C9orf72 [37], respectively.
-
Glutamate-mediated excitotoxicity has been suggested to cause motor neuron deterioration, and could be an indirect consequence of ALS-associated gene mutations such as in SOD1 or C9orf72, resulting in an elevated level of glutamate in the cerebrospinal fluid of patients [38][39][40].
-
RNA processing and metabolism is another key pathway affected in ALS. For example, mutations to RNA-binding proteins encoded by FUS, TDP-43, hnRNPA1, hnRNPA2B1, and MATR3, result in altered mRNA splicing, RNA nucleocytoplasmic transport and translation [18][41][42][43][44][45][46], as well as in the generation and accumulation of toxic stress granules [47]. Similarly, accumulation of toxic RNA foci can be observed in motor neurons in the context of C9orf72 mutations, and may lead to the sequestration of splicing proteins, thus affecting RNA maturation and translation [48]. The biogenesis of microRNA is also directly affected by mutated FUS, TDP-43, or C9orf72-mediated DPRs, thus having an impact on the expression of genes involved in motor neuron survival [18][49].
Understanding the functional processes that drive ALS pathology has proven to be a difficult and complex task, compounded by the heterogeneity that characterises the disease. The gene products of the 30 or more known ALS-associated genes interact with each other, are implicated in multiple molecular pathways, and result in multiple disease phenotypes, making functional curation and interpretation complex [9][18]. In addition, these monogenic causes in ALS occur only in ∼15% of sporadic ALS and ∼66% of familial ALS patients, so that more than 80% of the ALS population do not currently have any known ALS-associated mutations [9][12]. Nonetheless, acquiring an in-depth understanding of the molecular mechanisms and the genetic architecture of ALS could potentially lead to the identification of multiple patient strata and therefore targeted therapies to be applied to different subgroups of ALS patients.
2. ALS Genome-Wide Association Studies
In recent years, advances in high-throughput technologies have enabled the discovery of multiple Single Nucleotide Polymorphisms (SNPs) that are associated with ALS, mainly by the application of the Genome-Wide Association Study (GWAS) approach. GWAS aims to identify SNPs and other types of genetic variation (such as structural variants, copy number variations and multiple nucleotide polymorphisms) that are more frequent in patients than in people without the disease [50]. Statistical tests are carried out for disease association across genetic markers numbering from hundreds of thousands up to millions, depending on the genomic analytical platform. The most popular genotype-phenotype association studies use statistical models such as logistic or linear regression, depending on whether the trait is binary (i.e., case-control studies, such as ALS versus healthy controls) or quantitative (e.g., different scales of height). GWAS has been successful in discovering tens of thousands of significant genotype-phenotype associations in a large spectrum of diseases and traits, such as schizophrenia, anorexia nervosa, body-mass index (BMI), type 2 diabetes, and ALS [11][51][52][53]. Over the past decade, the discovery of significant genotype-phenotype associations has provided new insights into disease susceptibility, pathology, prevention, drug design and personalized medical approaches [52][54][55].
So far, numerous ALS GWAS studies have been published, aiming to identify novel ALS-associated variants through standard genotype-phenotype analyses. The first was published in 2007, providing genomic data for 276 cases and 271 controls [56]. Rapid recent technological advances and great efforts in the field have led to the genomic profiling of large ALS cohorts, providing new insights into the pathology of ALS [11]. The largest release of ALS genomic data was published in 2018 by Nicolas et al., and identified KIF5A as a novel ALS-associated gene; the study included a publicly-available large meta-analysis dataset of 10,031,630 imputed SNPs of 20,806 ALS and 59,804 controls as well as providing controlled access to “raw” genomic data including SNP-arrays of 12,188 cases and 3,292 controls [11][57]. Initiatives such as Project MinE and dbGaP have contributed to the systematic release of ALS GWAS data [57][58]. The ALSoD publicly available database for genes that are implicated in ALS records 126 genes, with a subset having been reproduced in multiple studies [59]. As of July 2020, the GWAS Catalogue has published 317 variants and risk allele associations with ALS [51].
2.1. The Genetic Architecture of ALS
The genetic contribution to familial and sporadic ALS has not been fully explained by genotype-phenotype discoveries [8][16], and the known Mendelian causes of ALS represent only a small proportion of the ALS population [9][12]. Nonetheless, estimates of heritability are high in sporadic ALS patients - for example, 61% in a twin meta-analysis study - suggesting that genetic factors are strongly represented in sporadic ALS and that further investigation may yet identify novel causal variants and/or multilocus interactions that could account for this high estimated heritability [60].
So far, evidence supports a model implicating rare variants (minor allele frequency <1%) along with non-genetic causes, such as environmental factors [3][27][61][62]. Large GWAS efforts suggest a genetic architecture for ALS that falls somewhere in the middle of the spectrum of genetic pathology in terms of effect size and prevalence of risk variants-i.e., an intermediate genetic architecture, lying between conditions such as schizophrenia which have many common variants each imparting a small increase to disease risk, and conditions such as Huntington’s disease which are caused by rare large-effect variants located in a single gene [3][10][63][64].
Many ALS-associated variants, particularly for C9orf72, also contribute to other conditions such as frontotemporal dementia (FTD) and cerebellar disease, suggesting that ALS is a multi-system syndrome [3][61][62]. ALS has an established overlap with other neurodegenerative and neuropsychiatric disorders, investigation of which could lead to insights into the understanding of pathology [3][5][16][61][65][66]. An example of this is the degree of overlap between familial ALS (∼40%) and familial FTD (∼25%) patients that carry the G6C4 expansion of C9orf72 [66][67]. C9orf72 hexanucleotide expansion has been associated to multiple traits including Alzheimer’s and Parkinson’s diseases, ataxia, chorea and schizophrenia [12][68][69][70]. A population-based GWAS study reported a higher prevalence of psychosis, suicidal behaviour, and schizophrenia, in Irish ALS kindreds, which was associated with the C9orf72 repeat expansion, based on an aggregation analysis [65]. Further evidence for a shared susceptibility to ALS was provided by the greater occurrence of dementia among first-degree relatives of ALS patients [70]. Several studies have suggested that the genetic overlap between ALS and other neurodegenerative and neuropsychiatric disorders could also be explained by the presence of ALS-associated pleiotropic variants that influence multiple, and in some cases quite distinct, phenotypic traits [71][72][73]. One study that supports this hypothesis is that of O’Brien et al., which shows that first-degree and second-degree relatives of Irish ALS patients have a significantly higher prevalence of schizophrenia and neuropsychiatric diseases than healthy controls, including obsessive-compulsive disorder, psychotic illness, and autism-the authors performed k-means clustering and calculated the relative risk to estimate aggregation [72][74][75][76].
3. ALS-Specific GWAS Challenges and Limitations
Despite that hundreds of ALS-associated variants have been recorded in public databases such as the GWAS Catalog [51], these associations show very little reproducibility across different studies and have not been able to explain a large percentage of ALS heritability [3][27]; a phenomenon which is generally known as the “missing heritability” paradox [77]. It has been proposed that SNPs contribute ∼8.5% of the overall heritability of ALS, although it should be noted that such estimates consider only linear single-marker effects of SNPs [27][77]. Here we outline some general GWAS limitations in the context of ALS, as well as potential reasons why standard GWAS phenotype-genotype analysis is unlikely to fully explain the genetic architecture of ALS.
A first general challenge in large scale genomic analyses is to ensure a high quality of the genotype data, so that the downstream results of the experimental design reflect true biology and not artifacts. Therefore, the collected genomic data first need to pass a comprehensive Quality Control (QC) pipeline including multiple sample and variant QC steps [78][79][80][81]. One challenge is that each dataset has its own specific features, thus there are not fixed thresholds for each quality-control step. For this reason, each study needs to follow a data-driven approach, taking into consideration the distribution of each data metric. However, there are some good practices in QC that may be generally applicable to most studies [78][81]. For example, it is typical to follow a procedure first filtering out low quality samples then removing poor quality markers, the order of this ensuring that as many genetic markers as possible are kept in the final dataset. However, overly strict thresholds can lead to the loss of a substantial proportion of samples, reducing study power. Another challenge is to ensure homogeneity of the collected samples in terms of ancestry. This QC step is carried out by analysing the population structure to remove ethnic outliers, and by accounting for confounding factors in later stages of the analysis, such as a potential inner population sub-structure, usually using the first few Principal Components, after performing a Principal Component Analysis on the homogeneous sample cohort. Also, it is very important to check for duplicated samples and, in non-family GWAS analyses, ensure that all samples are unrelated so that specific genotypes are not over-represented (and thereby contributing a bias to the subsequent analysis). Identity-by-descent (IBD) is a metric that corrects for such bias and takes into account the number of variants that a pair of individuals share.
GWAS is a single marker analysis treating each variant association as an independent event that contributes to the phenotype. Due to this, it is a standard practice for results to be corrected under the strict multiple testing threshold (p < 5 × 10) of the Bonferroni correction in order to control for false positive discoveries (Family-wise type I errors). This threshold derives from the hypothesis of 1,000,000 independent markers being tested under a significance level of 5%. Particularly in low sample size studies this correction can result in a loss of power of the analysis, which may then fail to capture a portion of potential risk variants that do not pass the significance threshold (Family-wise type II errors) [55][82].
Univariate analyses such as GWAS that test trait association for one locus at a time are not able to capture multilocus interactions-a phenomenon called epistasis-and the interaction of the environment with the genome; events that could potentially account for the missing heritability of ALS and explain the disease pathology [83][84]. The term epistasis was introduced in genetics over a century ago by Bateson et al. [85], and genetic and evolutionary biology studies have highlighted the importance of gene-gene interactions not only in the genetic architecture of an organism but also in evolution [77][86][87]. Epistasis represents non-additive events in the genome including interactions among two or more loci that have an effect on the phenotype [88]. Several studies have highlighted the role of epistasis in pathology, showing that SNP interactions provide a stronger association to the disease than the participating SNPs do individually [77][84][89][90]. To understand pathology in a complex disease such as ALS, it may be necessary to identify complex genetic interactions, including epistatic interations [77][87]. Nevertheless, the study of multilocus interactions poses a number of challenges, in particular the need for a high computational power as the number of tested interactions is extremely high even in pairwise combinations. As such, multivariate computational approaches and appropriate machine learning methods may be able to capture the potentially complex relationships among risk variants in ALS [77][90][91].
GWAS is more successfully employed under a “common disease-common variant” hypothesis, being of particular use in common diseases such as schizophrenia which are driven by many risk alleles each with high frequency [92]. In contrast, ALS is a heterogeneous disease likely comprised of multiple strata each resulting from combinations of different rare mutations and other factors. As a result, stratum-specific mutations may each have very small effects that are diluted and thus not captured by GWAS [3][27]. The majority of GWAS analyses have used SNP-arrays as they have until recently had a lower experimental cost in comparison to sequencing of the exome or the whole genome. SNP-array analyses can typically capture the effect of only common variants to the phenotype whereas sequencing analyses identify both common and rare variants. In most SNP-array GWAS studies, variants with Minor Allele Frequency (MAF) of <1–5% are removed from subsequent analysis as they are generally more difficult to genotype and therefore are considered potential false positives [55][78]. Nevertheless, whole genome sequencing, custom designed exome sequencing arrays, rare variant burden analyses and imputation approaches using large reference panels (such as the Haplotype Reference Consortium, which contains 64,976 haplotypes), face this challenge by recovering both rare (up to 0.1% MAF) and common variants that SNP-array platforms do not usually contain [55][93][94][95]. However, there is still a proportion of low frequency minor allele effects on the phenotype that cannot yet be detected by GWAS approaches and that could also potentially explain some of the missing heritability in ALS [3][27].
Lastly, another common GWAS challenge in complex diseases is the difficulty to distinguish causal variants from other non-disease-associated variants that are in high linkage disequilibrium [55]. Linkage disequilibrium describes the phenomenon where an allele of a variant is inherited together with the alleles of other variants [50]. These alleles of other variants are highly correlated and will have very similar GWAS signals with the truly causal SNP. The majority of disease-related variants are located in cis-regulatory regions of the genome [96], and given our limited knowledge of non-coding genomic loci, it is even more challenging for those to discern causal SNPs from the noise. Our difficulty to identify the causal variants in complex diseases among a pool of statistically significant associated variants adds to the challenge of identifying molecular processes that could have a significant impact on the disease.
Advanced machine learning prediction models trained in ALS genomic data could overcome the aforementioned challenges, moving towards better insights into disease causality and ultimately to a personalized understanding of ALS [55][97]. In Figure 1, we describe the basic steps of an ALS machine learning experimental design in order to discover ALS-associated novel loci or combinations of loci, as well as the main challenges of each step. Each of the main challenges is addressed in successive chapters of our review [98], as we describe and compare the experimental design of collected gene prioritization studies. Some of the challenges in Figure 1 have already been mentioned, such as the need for a large sample size that could increase the power of the study, a comprehensive quality control pipeline to assure high quality genomic data, as well as the curse of dimensionality which is a very common problem in genomic studies that include an extremely high number of features and especially in studies that focus on multilocus interactions.
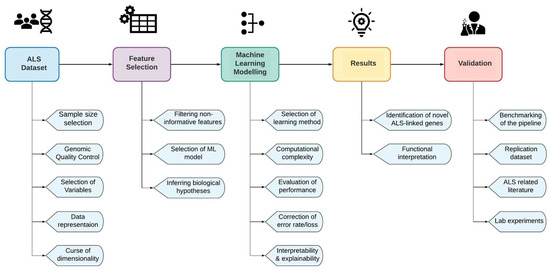
Figure 1. The main challenges of an ALS machine learning experimental design.
References
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408.
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130.
- Al-Chalabi, A.; Van Den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96.
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 1–6.
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700.
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323.
- Nicaise, C.; Mitrecic, D.; Pochet, R. Brain and spinal cord affected by amyotrophic lateral sclerosis induce differential growth factors expression in rat mesenchymal and neural stem cells. Neuropathol. Appl. Neurobiol. 2011, 37, 179–188.
- McLaughlin, L.R.; Vajda, A.; Hardiman, O. Heritability of amyotrophic lateral sclerosis insights from disparate numbers. Jama Neurol. 2015, 72, 857–858.
- Vijayakumar, U.G.; Milla, V.; Stafford, M.Y.C.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.R. A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front. Neurol. 2019, 10, 400.
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3.
- Nicolas, A.; Kenna, K.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.
- Turner, M.R.; Al-Chalabi, A.; Chio, A.; Hardiman, O.; Kiernan, M.C.; Rohrer, J.D.; Rowe, J.; Seeley, W.; Talbot, K. Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 2017, 88.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102.
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258.
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549.
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58.
- Morgan, S.; Duguez, S.; Duddy, W. Personalized Medicine and Molecular Interaction Networks in Amyotrophic Lateral Sclerosis (ALS): Current Knowledge. J. Pers. Med. 2018, 8, 44.
- Gall, L.L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duguez, S. Molecular and cellular mechanisms affected in ALS. J. Pers. Med 2020, 10, 101.
- Volonté, C.; Morello, G.; Spampinato, A.G.; Amadio, S.; Apolloni, S.; D’Agata, V.; Cavallaro, S. Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res. Rev. 2020, 62, 101121.
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, 1005357.
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro 2017, 4, 249–265.
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075.
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L.G. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849.
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62.
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 2015, 47, 834–841.
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135.
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; Van Der Spek, R.A.; Võsa, U.; De Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048.
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162.
- De vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; Mcloughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728.
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456.
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 1–10.
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death. Neuron 2014, 84, 1213–1225.
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Greg Stacey, R.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759.
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal exocytosis, exosome release and secretory autophagy: The autophagic- and endo-lysosomal systems go extracellular. Int. J. Mol. Sci. 2020, 21, 2576.
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077.
- Blanc, L.; Vidal, M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases 2018, 9, 95–106.
- Laslo, P.; Lipski, J.; Nicholson, L.F.; Miles, G.B.; Funk, G.D. GluR2 AMPA Receptor Subunit Expression in Motoneurons at Low and High Risk for Degeneration in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2001, 169, 461–471.
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78.
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042.
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695.
- Buratti, E.; Baralle, F.E. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator of CFTR Exon 9. J. Biol. Chem. 2001, 276, 36337–36343.
- Leblond, C.S.; Gan-Or, Z.; Spiegelman, D.; Laurent, S.B.; Szuto, A.; Hodgkinson, A.; Dionne-Laporte, A.; Provencher, P.; de Carvalho, M.; Orrù, S.; et al. Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2016, 37, 17–209.
- Jutzi, D.; Akinyi, M.V.; Mechtersheimer, J.; Frilander, M.J.; Ruepp, M.D. The emerging role of minor intron splicing in neurological disorders. Cell Stress 2018, 2, 40–54.
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19.
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666.
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688.
- Kumar, V.; Hasan, G.M.; Hassan, M.I. Unraveling the role of RNA mediated toxicity of C9orf72 repeats in C9-FTD/ALS. Front. Neurosci. 2017, 11, 711.
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352.
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-Wide Association Studies. PLoS Comput. Biol. 2012, 8, e1002822.
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901.
- Klein, R.J.; Xu, X.; Mukherjee, S.; Willis, J.; Hayes, J. Successes of Genome-wide association studies. Cell 2010, 142, 350–351.
- Zhao, W.; Rasheed, A.; Tikkanen, E.; Lee, J.J.; Butterworth, A.S.; Howson, J.M.; Assimes, T.L.; Chowdhury, R.; Orho-Melander, M.; Damrauer, S.; et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat. Genet. 2017, 49, 1450–1457
- Duncan, L.; Yilmaz, Z.; Gaspar, H.; Walters, R.; Goldstein, J.; Anttila, V.; Bulik-Sullivan, B.; Ripke, S.; Thornton, L.; Hinney, A.; et al. Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. Am. J. Psychiatry 2017, 174, 850–858.
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484.
- Schymick, J.C.; Scholz, S.W.; Fung, H.C.; Britton, A.; Arepalli, S.; Gibbs, J.R.; Lombardo, F.; Matarin, M.; Kasperaviciute, D.; Hernandez, D.G.; et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2007, 6, 322–328.
- Mailman, M.D.; Feolo, M.; Jin, Y.; Kimura, M.; Tryka, K.; Bagoutdinov, R.; Hao, L.; Kiang, A.; Paschall, J.; Phan, L.; et al. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007, 39, 1181–1186.
- Van Rheenen, W.; Pulit, S.L.; Dekker, A.M.; Al Khleifat, A.; Brands, W.J.; Iacoangeli, A.; Kenna, K.P.; Kavak, E.; Kooyman, M.; McLaughlin, R.L.; et al. Project MinE: Study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 2018, 26, 1537–1546.
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351.
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326.
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782.
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615.
- Myers, R.H. Huntington’s Disease Genetics. NeuroRx 2004, 1, 255–262.
- Loh, P.R.; Bhatia, G.; Gusev, A.; Finucane, H.K.; Bulik-Sullivan, B.K.; Pollack, S.J.; Lee, H.; Wray, N.R.; Kendler, K.S.; O’donovan, M.C.; et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015, 47, 1385.
- Byrne, S.; Heverin, M.; Elamin, M.; Bede, P.; Lynch, C.; Kenna, K.; MacLaughlin, R.; Walsh, C.; Al Chalabi, A.; Hardiman, O. Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: A population-based case-control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 699–708.
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23.
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330.
- Majounie, E.; Abramzon, Y.; Renton, A.E.; Perry, R.; Bassett, S.S.; Pletnikova, O.; Troncoso, J.C.; Hardy, J.; Singleton, A.B.; Traynor, B.J. Repeat expansion in C9ORF72 in Alzheimer’s disease. N. Engl. J. Med. 2012, 366, 283.
- Lesage, S.; Le Ber, I.; Condroyer, C.; Broussolle, E.; Gabelle, A.; Thobois, S.; Pasquier, F.; Mondon, K.; Dion, P.A.; Rochefort, D.; et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013, 136, 385–391.
- Majoor-Krakauer, D.; Ottman, R.; Johnson, W.G.; Rowland, L.P. Familial aggregation of amyotrophic lateral sclerosis, dementia, and Parkinson’s disease: Evidence of shared genetic susceptibility. Neurology 1994, 44, 1872–1877.
- Karch, C.M.; Wen, N.; Fan, C.C.; Yokoyama, J.S.; Kouri, N.; Ross, O.A.; Höglinger, G.; Müller, U.; Ferrari, R.; Hardy, J.; et al. Selective genetic overlap between amyotrophic lateral sclerosis and diseases of the frontotemporal dementia spectrum. JAMA Neurol. 2018, 75, 860–875.
- O’Brien, M.; Burke, T.; Heverin, M.; Vajda, A.; McLaughlin, R.; Gibbons, J.; Byrne, S.; Pinto-Grau, M.; Elamin, M.; Pender, N.; et al. Clustering of neuropsychiatric disease in first-degree and second-degree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2017, 74, 1425–1430.
- Stearns, F.W. One hundred years of pleiotropy: A retrospective. Genetics 2010, 186, 767–773.
- Wu, J.; Liu, H.; Xiong, H.; Cao, J.; Chen, J. K-means-based consensus clustering: A unified view. IEEE Trans. Knowl. Data Eng. 2015, 27, 155–169.
- Wagstaff, K.; Cardie, C.; Rogers, S.; Schroedl, S. Constrained K-means Clustering with Background Knowledge. Proc. Eighteenth Int. Conf. Mach. Learn. 2001, 1, 577–584.
- MacQueen, J.B. Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1967; pp. 281–297.
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198.
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, P.; Zondervan, K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2011, 5, 1564–1573.
- Marees, A.T.; de Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, 1–10.
- Verma, S.S.; de Andrade, M.; Tromp, G.; Kuivaniemi, H.; Pugh, E.; Namjou-Khales, B.; Mukherjee, S.; Jarvik, G.P.; Kottyan, L.C.; Burt, A.; et al. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 2014, 5, 1–15.
- Laurie, C.C.; Doheny, K.F.; Mirel, D.B.; Pugh, E.W.; Bierut, L.J.; Bhangale, T.; Boehm, F.; Caporaso, N.E.; Cornelis, M.C.; Edenberg, H.J.; et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol. 2011, 34, 591–602.
- Dudbridge, F.; Gusnanto, A. Estimation of significance thresholds for genomewide association scans. Genet. Epidemiol. 2008, 32, 227–234.
- Culverhouse, R.; Suarez, B.K.; Lin, J.; Reich, T. A perspective on epistasis: Limits of models displaying no main effect. Am. J. Hum. Genet. 2002, 70, 461–471.
- Hemani, G.; Knott, S.; Haley, C. An Evolutionary Perspective on Epistasis and the Missing Heritability. PLoS Genet. 2013, 9, 1003295.
- Bateson, W.; Saunders, E.; Punnett, R.; Sons, C.H.U.H. Reports to the Evolution Committee of the Royal Society, Report II. London. R. Soc. 1905, 2, 5–131.
- de Visser, J.A.G.M.; Cooper, T.F.; Elena, S.F. The causes of epistasis. Proc. R. Soc. B Biol. Sci. 2011, 278, 3617–3624.
- Pan, Q.; Hu, T.; Moore, J.H. Epistasis, Complexity, and Multifactor Dimensionality Reduction; Humana Press: Totowa, NJ, USA, 2013; pp. 465–477.
- Churchill, G.A. Epistasis. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 505–507.
- Goudey, B.; Rawlinson, D.; Wang, Q.; Shi, F.; Ferra, H.; Campbell, R.M.; Stern, L.; Inouye, M.T.; Ong, C.S.; Kowalczyk, A. GWIS–model-free, fast and exhaustive search for epistatic interactions in case-control GWAS. BMC Genom. 2013, 14 (Suppl. S3), 1–18.
- Yin, B.; Balvert, M.; Van Der Spek, R.A.; Dutilh, B.E.; Bohté, S.; Veldink, J.; Schönhuth, A. Using the structure of genome data in the design of deep neural networks for predicting amyotrophic lateral sclerosis from genotype. Bioinformatics 2019, 35, i538–i547.
- Kim, N.C.; Andrews, P.C.; Asselbergs, F.W.; Frost, H.R.; Williams, S.M.; Harris, B.T.; Read, C.; Askland, K.D.; Moore, J.H. Gene ontology analysis of pairwise genetic associations in two genome-wide studies of sporadic ALS. BioData Min. 2012, 5, 9.
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. 2001, 17, 502–510.
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283.
- Naj, A.C. Genotype Imputation in Genome-Wide Association Studies. Curr. Protoc. Hum. Genet. 2019, 102, e84.
- Pistis, G.; Porcu, E.; Vrieze, S.I.; Sidore, C.; Steri, M.; Danjou, F.; Busonero, F.; Mulas, A.; Zoledziewska, M.; Maschio, A.; et al. Rare variant genotype imputation with thousands of study-specific whole-genome sequences: Implications for cost-effective study designs. Eur. J. Hum. Genet. 2015, 23, 975–983.
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195.
- Myszczynska, M.A.; Ojamies, P.N.; Lacoste, A.M.; Neil, D.; Saffari, A.; Mead, R.; Hautbergue, G.M.; Holbrook, J.D.; Ferraiuolo, L. Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nat. Rev. Neurol. 2020, 16, 440–456.
- Vasilopoulou, Christina; Morris, Andrew P.; Giannakopoulos, George; Duguez, Stephanie; Duddy, William; What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis?. J. Pers. Med. 2020, 10, 247, https://doi.org/10.3390/jpm10040247.


