 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudio Luparello | + 3287 word(s) | 3287 | 2020-11-30 07:00:14 | | | |
| 2 | Vivi Li | -27 word(s) | 3260 | 2020-12-10 05:03:52 | | | | |
| 3 | Claudio Luparello | + 1 word(s) | 3261 | 2020-12-11 09:45:46 | | |
Video Upload Options
Histone deacetylases (HDACs) are key components of the epigenetic machinery controlling gene expression. They are involved in chromatin remodeling events via post-translational histone modifications but may also act on nonhistone proteins, influencing many fundamental cellular processes. Due to the key involvement of HDACs in serious human pathologies, including cancer, HDAC inhibitors (HDACis) have received increased attention in recent years. It is known that marine invertebrates produce significant amounts of secondary metabolites showing active pharmacological properties and an extensive spectrum of biomedical applications. Some of these compounds possess HDACi properties.
1. Histone Deacetylase Inhibitors
Histone deacetylases (HDACs) are a key group of enzymes, highly conserved throughout evolution, involved in chromatin remodeling events via post-translational histone modification by catalyzing the removal of acetyl groups from lysine residues in their amino termini, thus determining chromatin condensation and transcriptional repression. These enzymes ay also act on non-histone proteins, thereby influencing many fundamental cellular processes, including microtubule dynamics and intracellular transport, metabolism, and aging [1],[2],[3]. HDACs are involved in cancer-associated cell hyperproliferation, invasion, and metastasis [4],[5].
Histone deacetylase inhibitors (HDACis) belong to a large group of chemically different compounds, which includes complex molecules such as hydroxamates, cyclic tetrapeptides/epoxides, and benzamides, and relatively simple short-chain aliphatic acids, such as valproic acid and butyric acid [6],[7],[8],[9],[10],[11]. This chemical heterogeneity implies a difference in the precise molecular mechanism of action that, for several members, is still poorly understood and, also, given that eighteen diverse HDACs exist in mammalian cells, in the differing intracellular localizations, interactions with other proteins, and associations in multimolecular complexes.
HDACis are well-known antineoplastic compounds, the most potent to date being trichostatin A, active in vitro at nanomolar concentrations, which is a fermentation product of Streptomyces bacteria originally utilized as an antimycotic agent and later found to restrain the proliferation of cultured tumor cells [12]. The need for alternative HDACis, due to the difficulties and costs related to trichostatin A production, determined the introduction of a number of other inhibitors, such as butyrate, phenylbutyrate, depsipeptide, pyroxamide, suberoylanilide bishydroxyamide, valproic acid, and N-acetyldinaline in the clinical trials for anticancer activity, thus expanding the opportunities for epigenetic therapies [13]. In addition to the antineoplastic mechanism based upon reprogramming of the gene expression that ultimately suppresses cell proliferation and motility, HDACis have been proven to promote tumor cell death via apoptosis in a more powerful way when used in combination with other agents, e.g., [[14]], and, also, to inhibit endothelial cell growth and tumor angiogenesis [15],[16]. Noteworthy, further applications of these compounds currently under study include the treatment of fungal infections and human neurological pathologies, such as Rett syndrome, Friedrich’s ataxia, Huntington’s chorea, and spinal muscular atrophy [17],[18],[19],[20],[21].
Seas and oceans, which cover three-quarters of the globe, host an enormous diversity of organisms, many of which are still unknown, and represent the underexploited richest source of bioactive marine natural products. Invertebrates, which account for more than 50% of the species colonizing the aquatic environment in Europe, are important environmental bioindicators [22],[23],[24],[25]. They are known to produce significant amounts of secondary metabolites with unique chemical skeletons as molecular cues addressed to regulate very disparate biological activities, e.g., feeding; inter- and intraspecific signalizations; mating; predation; and defense from predators, competitors, pathogenic microorganisms, and UV radiation damage. These products contribute to the adaptation mechanisms to the specific life conditions in the greatly different marine ecosystems [26]. A large proportion of invertebrate-derived extracts and isolated compounds has shown active pharmacological properties, such as anticancer, antimicrobial, and anti-inflammatory, among others, with an extensive spectrum of biomedical applications that makes them already approved or prospective drugs of marine origin with promising results for different therapeutic purposes [27],[28],[29],[30],[31],[32]. Within this scenario, the aim of this entry is to summarize the information on the marine invertebrate-derived chemicals that possess HDACi properties that will be dealt with according to the taxonomic hierarchy of the producing invertebrate species. In particular in some species of invertebrates belonging to the phyla of Porifera, Cnidaria and Echinodermata, the presence of compounds with HDAC1 properties has been highlighted.
2. Porifera
Porifera is the oldest still-existing metazoan group, which comprises sponges, sessile aquatic organisms endowed with a very simple pattern of body organization constituted by a skin covering a collagenous matrix crossed by canals and microscopic chambers. Several species of porifera have been useful for isolating and characterizing compounds with HDACi activities such as: Aplysinella rhax (Laubenfels, 1954; Demospongiae, Verongiida: Aplysinellidae), originally described as Dysidea rhax [33]; Psammaplysilla purpurea, currently Pseudoceratina purpurea (Carter, 1880; Demospongiae, Verongiida: Pseudoceratinidae); Dendrilla lacunosa, currently Ernstilla lacunosa (Hentschel, 1912; Demospongiae, Dendroceratida: Darwinellidae); Theonella swinhoei (Gray, 1868; Demospongiae, Tetractinellida: Theonellidae); Petrosia alfiani (de Voogd and van Soest, 2002; Demospongiae, Haplosclerida: Petrosiidae) and some species of the genus Jaspis (Gray, 1867; Demospongiae, Tetractinellida: Ancorinidae) Halichondria (Fleming, 1828; Demospongiae, Suberitida: Halichondriidae); Haliclona (Demospongiae, Haplosclerida: Chalinidae) and Xestospongia (Demospongiae, Haplosclerida: Petrosiidae), in particular Xestospongia vansoesti (Bakus and Nishiyama, 2000)
2.1. Psammaplins
Other members of the psammaplin group, i.e., psammaplin B–J and bisaprasin, were subsequently isolated and characterized, but only psammaplins A and F, the latter differing for a C2H2NO3 terminal group, a carboxylic acid moiety, a secondary amide functional group, and an N-substituted oxalamic acid group, and bisaprasin (Figure 1B) were proven to have HDACi properties when incubated with the 3H-acetylated human histone H4 peptide substrate [35],[38].
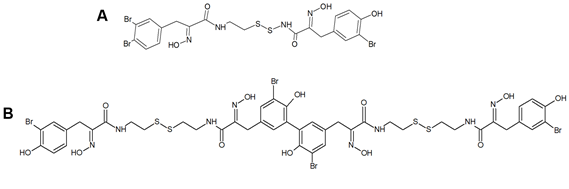
Figure 1. Structures of psammaplin A (A) and bisaprasin (B)
In the following lines, the effects of psammaplins on neoplastic cell model systems are recapitulated.
Psammaplins and breast cancer cells: In light of molecular modeling data identifying several potential-binding sites for psammaplin A within the peroxisome proliferator-activated receptor γ (PPARγ) ligand-binding pocket, Mora et al. [39] demonstrated the psammaplin-induced activation of the receptor in a MCF-7 breast tumor cell-based reporter assay, followed by the promotion of apoptotic death at least in part mediated by the switch-on of the PPARγ-regulated gene expression. In 2012, Baud et al. [40] demonstrated the HDAC isoform selectivity of psammaplin A, which appeared to be 360-fold selective for HDAC1 over HDAC6 and more than 1000-fold less powerful towards HDAC7 and HDAC8, being the same selectivity profile maintained in MCF-7 cells as an in vitro model system. More recently, Zhou et al. [41] exposed both the estrogen-dependent T47D cells and different genetically characterized metastatic subclones of triple-negative MDA-MB231 cells specific to lung, bone, and brain to different psammaplins. They found a powerful inhibitory activity of the proliferation and three-dimensional invasive growth of tumor cells, except for the brain metastatic subclone, by the stereo isomers (e,z)-psammaplin A and (e,e)-psammaplin A compared to bisaprasin and psammaplins E and K. From a molecular point of view, psammaplin A was proven to trigger the activity of the hypoxia-inducible factor (HIF) and the upregulation of HIF target genes, such as cyclin-dependent kinase inhibitor 1A (CDKN1A) and vascular endothelial growth factor A (VEGFA; this only by the e,e isomer) and the downregulation of sirtuin-1 (SIRT1), the latter leading to the increased p53 acetylation and autophagy-related gene expression and, ultimately, to autophagic cell death.
Psammaplin A and endometrial cancer cells: Ahn et al. [42] reported the inhibitory effect of the compound on Ishikawa endometrial cancer cells via cell cycle arrest at the G1 and G2/M phases and apoptosis promotion. Molecular events associated with the impairment of the cell cycle and the induction of apoptosis were the downregulation of cyclin D1, cyclin E, and CDK4 (involved in the block of G1 phase progression); cyclin A, cyclin B1, and CDK2 (involved in the block of G2/M phase progression); and the upregulation of p21WAF1, along with a decrease in the level of hyperphosphorylated pRb.
Psammaplin A and C and glioblastoma cells: psammaplin C (Figure 2) is a powerful inhibitor of carbonic anhydrase XII [43], whose activity is required to ensure an efficient efflux of chemotherapeutics by a P-glycoprotein pump in tumor cells. Salaroglio et al. [44] reported that carbonic anhydrase XII is overexpressed in glioblastoma stem cells and that a combination of psammaplin C and temozolomide, the latter being the first-line drug in glioblastoma treatment, rescues its efficacy against the highly chemorefractory stem component of the glioblastoma cell population. More recent data [45] demonstrated an even greater efficacy, in the order of subnanomolar carbonic anhydrase XII inhibitory activity, by a variant of the molecule endowed with a thiadiazole sulfonamide moiety replacing the ethyl sulfonamide one, which was able to inhibit also carbonic anhydrase IX, the other cancer-associated isozyme.
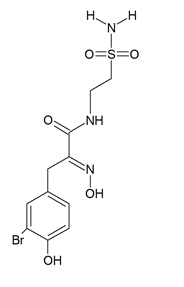
Figure 2. Structure of psammaplin C
Dealing with psammaplin A, a molecular study performed by Ratovitski [46] on U87-MG glioblastoma cells demonstrated the ability of the compound to induce the expression and phosphorylation of TP53 family members instrumental for the transcriptional activation of downstream target genes such as those involved in autophagy signaling, i.e., coding for the Autophagy-Related 5 protein and UVRAG coding for the UV Radiation Resistance-Associated protein. Confirmation of the autophagy flux-promoting activity was obtained by an electrophoretic analysis of the LC3B-I/LC3B-II shift.
It must be mentioned that a number of experimental investigations, such as that of Mujumdar et al. [43] referenced above, have been focused on the synthesis and characterization of a series of psammaplin derivatives that might show a greater HDAC inhibitory efficacy and cytotoxic potential, although they provided mixed results. Among the most successful ones, the studies of Baud et al. [40] demonstrated the particularly potent activity of the (2E,2′E)-N,N′-(2,2′-Disulfanediylbis (ethane-2,1-diyl))bis(3-(3-bromo-4-methoxyphenyl)-2-(hydroxyimino)propanamide) variant towards MCF-7 breast cancer and A549 lung cancer cells. In addition, Byun et al. [47] reported the significant in vitro and in vivo anti-breast tumor and antimetastatic activity of psammaplin A-3091 (Figure 3), a heteromonomeric-structured analog with a tertiary butyl functional group able to specifically target DOT1L, coding for the disruptor of Telomeric silencing 1-like protein, involved in the regulation of both the epithelial-mesenchymal transition and the stem cell properties of breast cancer cells.
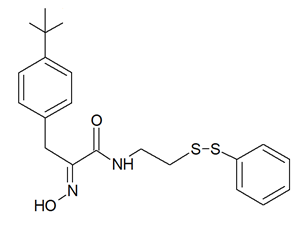
Figure 3. Structure of psammaplin A-3091 [47]
2.2. Yakushinamides
From the demosponge Theonella swinhoei (Gray, 1868; Demospongiae, Tetractinellida: Theonellidae, Figure 4) Takada et al. [48] isolated two prolyl amides of polyoxygenated fatty acid, i.e., yakushinamide A and B (Figure 4), which displayed a moderate inhibitory effect on HDACs and sirtuins. In particular, yakushinamide A inhibited HDAC1, SIRT1, and -3 at 26, 16, and 79 μM, respectively, whereas yakushinamide B at 29, 75, 52, 34, 150, and 78 μM, respectively.
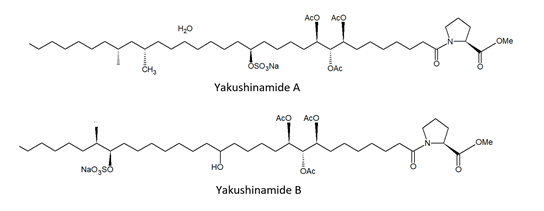
Figure 4. Structures of yakushinamide A and B
2.3. Halistanol sulphates
In the search for new SIRT inhibitors from marine sponges, from the species of the genus Halichondria (Fleming, 1828; Demospongiae, Suberitida: Halichondriidae) Nakamura et al. [49] isolated the highly sulphated steroid halistanol sulphate (Figure 5) and two novel analogs, i.e., halistanol sulphate I and -J, differing from the parental molecule for methylene protons and the cyclopropyl ring in the side chain, respectively. When submitted to an in vitro SIRT1-3 inhibitory assay, these compounds were shown to be active, with IC50 values of 45.9–67.9, 18.9–21.1, and 21.8–37.5 μM, respectively. On the other hand, they exerted no cytotoxic effects on HeLa cervical adenocarcinoma and P388 mouse leukemia cells at the concentration of 100 μM. Studies on the crystal structure of the SIRT3-halistanol sulphate complex indicated the existence of an allosteric site for enzyme inhibition far from the SIRT active site, substrate-binding site, and NAD+ cofactor-binding site, thus suggesting that steroid sulphates may be endowed with a good motif for the allosteric regulation of enzymes.
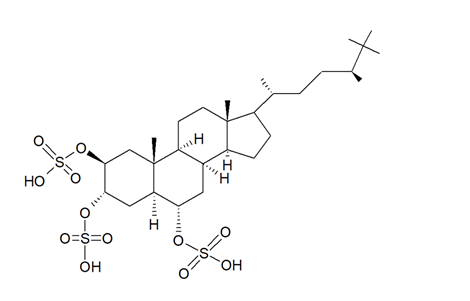
Figure 5. Structure of halistanol sulphate
2.4. Azumamides
From the species Mycale izuensis, Nakao et al. [50] isolated five cyclic tetrapeptides named azumamides A-E endowed with HDACi activity in enzymatic assays (Figure 6).
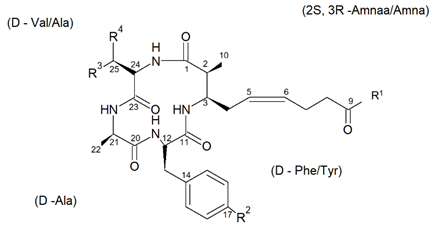
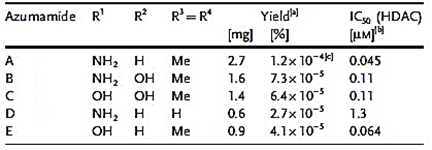
Figure 6. Structures of azumamides A-E. (a) Yield of azumamide isolated by extraction from a marine sponge. (b) Against the crude enzymes extracted from K562 cells. (c) Yield based on wet weight (from[50]).
Azumamide A was initially also tested at the cell level, showing a moderate cytostatic effect on K562 cells and a significant antiangiogenic effect on mouse vascular progenitor cells. Subsequently, a total synthesis was reported for the sole azumamides A and E, and research interest was mainly focused on the greater HDACi efficacy of azumamide E, which appeared able to inhibit total HDACs from HeLa cell extracts with a much lesser IC50 (110 ± 33 nM) with respect to that of azumamide A (5800 ± 1200 nM) and showed selectivity for HDAC1-4. In particular, from a biological point of view, the compound proved to be a strong inhibitor of in vitro angiogenesis by mouse-induced pluripotent stem cells [51],[52],[53]. More recently, the total synthesis of all five natural azumamides, as well as their profiling towards the whole panel of HDACs, has been reported. The data obtained revealed that azumamide C was a two-fold more potent inhibitor than azumamide E on the majority of HDAC isozymes. The observed discrepancy among the various evaluations of HDACi inhibitory activities was ascribed to the assay conditions largely affecting the results, underlining the need of their confirmation through parallel biological (e.g., antiproliferative or antiangiogenic) assays. On the other hand, surprisingly, given that the in vitro HDACi activity of azumamides B, C, and E were confirmed, no compound was able to reverse the chemoresistance caused by silencing of the proapoptotic Bim gene and, therefore, influence the growth of the Epstein−Barr virus (EBV)-infected human Burkitt’s lymphoma EB-3 cells differently from what was achieved upon treatment with HDACi SAHA [54],[55]. This prompts a future investigation on azumamide analogs designed on the original scaffold but endowed with a more potent and selective ligand activity.
2.5. Cyclostellettamines and dehydrocyclostellettamines
The species of the Haliclona and Xestospongia genus have proven to be good sources of natural HDACis. In 2004, Oku et al. [56] isolated cyclostellettamine A and three new cyclostellettamine alkaloids, i.e., cyclostellettamine G and dehydrocyclostellettamines D and E (Figure 7), from species of the genus Xestospongia, all displaying inhibitory activity with IC50 values in the range between 17 and 80 μM on HDAC preparations partially purified from K562 cells and exerting a moderate cytotoxic effect on HeLa human cervix carcinoma, P388 mouse leukemia, and 3Y1 rat fibroblastic cells.
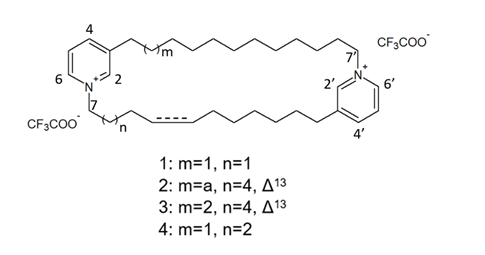
Figure 7. Structures of cyclostellettamine G (1), dehydrocyclostellettamine D (2), dehydrocyclostellettamine E, (3) and cyclostellettamine A (4) (from[56])
More recently, Lee et al. [57] isolated eight novel cyclic bis-1,3-dialkylpyridinium compounds, as well as the two cyclostellettamines N and Q, from a species of the genus Haliclona (Figure 8), which exhibited a moderate doxorubicin-comparable cytotoxicity towards A549 lung cancer cells and, also, a diverse range of antimicrobial activity specifically directed against Gram-positive bacterial strains.
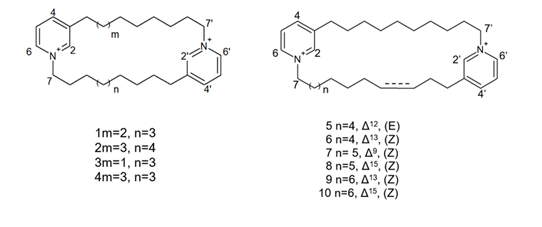
Figure 8. Structures of cyclostellettamine N (1), cyclostellettamine Q (2), and the other eight bis-1,3-dialkylpyridinium compounds (3–10) extracted from species of the genus Haliclona.
2.6. Halenaquinone
X. vansoesti and P. alfiani produce the polycyclic quinone-type metabolite halenaquinone (Figure 9) found to induce the inhibition of pan-HDACs and, in addition, also, topoisomerase IIα expression, the latter resulting in the switching-off of DNA replication.
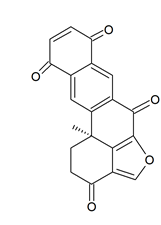
Figure 9. Structure of halenaquinone
Dealing with the biological aspects, Shih et al. [58] demonstrated its cytotoxic activity on a number of cancer cell lines, being particularly remarkable against Molt 4 leukemia cells, in which viability suppression and apoptosis promotion following reactive oxygen species (ROS)-induced mitochondrial dysfunction was observed. The molecular signatures associated to the exposure of leukemia cells to the molecule were the upregulation of cytochrome c of the proapoptotic protein Bax, of the cytosolic hexokinase I, and of the active forms of caspase 8 and -9 and the downregulation of the antiapoptotic proteins Bcl-2, Bid, and cytosolic hexokinase II and of the phosphorylated (activated) forms of Akt, phosphatase and tensin homolog (PTEN), glycogen synthase kinase 3 β (GSK3β), and phosphoinositide-dependent kinase-1 (PDK1). Of note, halenaquinone exhibited also a potent in vivo antileukemic effect in mice xenograft assays, reducing the weight and size of the tumor mass without affecting the total mice weight. In addition, Takaku et al. [59] demonstrated the inhibitory activity of the compound on DNA homologous pairing by direct binding to RAD51 enzyme, involved in the repair of DNA double-strand breaks, and subsequent likely competition with the double-strand DNA in the secondary DNA-binding site within the RAD51–single-strand DNA filament complex. More recently, Tsukamoto et al. [60] identified halenaquinone as an inhibitor of the receptor activator of nuclear factor-κB ligand (RANKL)-induced upregulation of tartrate-resistant acid phosphatase (TRAP), which is involved in cell fusion, leading to the formation of multinucleated osteoclasts.
3. Cnidaria
Xenia elongata (Dana, 1846; Anthozoa, Alcyonacea: Xeniidae) belongs to the cnidarians. This animal group constitutes the first metazoan phylum endowed with a well-developed degree of tissue organization and gathers polymorphic aquatic animals displaying radial or biradial symmetry and a single central body cavity (the “coelenteron”) with a mouth and tentacles but lacking an anus.
Andrianasolo et al. [61] isolated a new diterpene from X. elongata (Figure 10), which promoted the programmed death of immortalized apoptosis-competent W2 cells at a 1.2-μM concentration. Further enzymatic inhibition tests against the class I, -II A, and the -II B HDACs demonstrated that the compound specifically inhibited class II B HDAC6 with an IC50 of about 80 μM. In light of the data obtained, this molecule represents a new model structure of selective HDAC inhibitor that may be used for the development of novel HDAC isoform-targeting drugs.
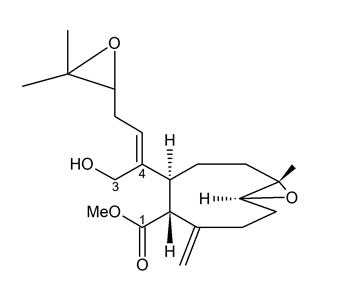
Figure 10. Structure of the diterpene extracted from X. elongata [61].
4. Echinodermata
Holopus rangii (Orbigny, 1837; Crinoidea, Cyrtocrinida: Holopodidae) belonging to the Echinoderms is a marine invertebrate endowed with a hard, spiny covering or skin. From this species Kemami Wangun et al. [62] isolated a novel member of the phenanthrol perylenequinone family of natural products denominated gymnochrome E (Figure 11) that showed an inhibitory activity towards purified HDAC-1 with an IC50 of 10.9 μM and selectively restrained the growth of the multidrug-resistant NCI/ADRRes ovarian cancer cell line with an IC50 value of 3.5 μM while exerting no effect on PANC-1 pancreatic carcinoma and DLD-1 human colorectal adenocarcinoma cell lines. Moreover, the compound appeared endowed with an antimicrobic activity against Staphylococcus aureus and its methicillin-resistant strain with a minimum inhibitory concentration of 25 μg/mL but not against Pseudomonas aeruginosa and Candida albicans.
Figure 11. Structure of gymnochrome E.
5. Conclusions
It is widely acknowledged that the enormous biodiversity of the marine habitats represents a source of immeasurable value for the isolation of very disparate bioactive secondary metabolites from bacteria, plants, and animals whose number is expected to increase rapidly, e.g., [63]. Among the enzymatic inhibitors searched in extracts from marine organisms, a focus has been put on HDACis for their wide range of biomedical applications. Natural inhibitors have been isolated from aquatic microorganisms [64],[65], and, as documented by the studies presented in this review, also, marine invertebrates have contributed with a number of compounds displaying impairing properties of various potency towards HDACs, demosponges being the most investigated to date. It is worth mentioning that some marine species-derived molecules showing structural similarities with known HDACis failed to exert HDACi-referable cellular and molecular effects, as in the case of SAHA- and trichostatin A-resembling N-(4-guanidinobutyl)-2-(4-hydroxy phenyl)-2-oxo-acetamide isolated from the cnidarian hydroid Campanularia sp. [66]. Therefore, a thorough biological characterization of the novel compounds identified in the future, including the identification of the specific, if any, target cytotype(s), appears to be a necessary aspect for the subsequent development of efficacious prevention and/or treatment agents against different pathological states—among them, cancers. On the other hand, the unique and peculiar chemical scaffolds presented by the marine-derived HDACis may be successfully utilized for the design of analogs with increased bioavailability and efficacy, less toxicity, and, also, very interestingly, higher isoform selectivity.
References
- Ibraheem Ali; Ryan J. Conrad; Eric Verdin; Melanie Ott; Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chemical Reviews 2018, 118, 1216-1252, 10.1021/acs.chemrev.7b00181.
- Kelly N. Hassell; Histone Deacetylases and their Inhibitors in Cancer Epigenetics. Diseases 2019, 7, 57, 10.3390/diseases7040057.
- Aikaterini F. Giannopoulou; Athanassios D. Velentzas; Eumorphia G. Konstantakou; Margaritis Avgeris; Stamatia A. Katarachia; Nikolaos C. Papandreou; Nikolas I. Kalavros; Vassiliki E. Mpakou; Vassiliki A. Iconomidou; Ema Anastasiadou; et al.Ioannis K. KostakisIssidora S. PapassideriGerassimos E. VoutsinasAndreas ScorilasDimitrios J. Stravopodis Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. International Journal of Molecular Sciences 2019, 20, 1291, 10.3390/ijms20061291.
- Nagendra K. Chaturvedi; Nathan D. Hatch; Garrett L. Sutton; Matthew Kling; Julie M. Vose; Shantaram S. Joshi; A novel approach to eliminate therapy-resistant mantle cell lymphoma: synergistic effects of Vorinostat with Palbociclib. Leukemia & Lymphoma 2018, 60, 1214-1223, 10.1080/10428194.2018.1520986.
- Mohammed I. Y. Elmallah; Olivier Micheau; Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers 2019, 11, 850, 10.3390/cancers11060850.
- Mariangela Librizzi; Alessandra Longo; Roberto Chiarelli; Jahanghir Amin; John Spencer; Claudio Luparello; Cytotoxic Effects of Jay Amin Hydroxamic Acid (JAHA), a Ferrocene-Based Class I Histone Deacetylase Inhibitor, on Triple-Negative MDA-MB231 Breast Cancer Cells. Chemical Research in Toxicology 2012, 25, 2608-2616, 10.1021/tx300376h.
- Mariangela Librizzi; Roberto Chiarelli; Liana Bosco; Supojjanee Sansook; Jose M. Gascon; John Spencer; Fabio CaraDonna; Claudio Luparello; The Histone Deacetylase Inhibitor JAHA Down-Regulates pERK and Global DNA Methylation in MDA-MB231 Breast Cancer Cells. Materials 2015, 8, 7041-7047, 10.3390/ma8105358.
- Mariangela Librizzi; John Spencer; Claudio Luparello; Biological Effect of a Hybrid Anticancer Agent Based on Kinase and Histone Deacetylase Inhibitors on Triple-Negative (MDA-MB231) Breast Cancer Cells. International Journal of Molecular Sciences 2016, 17, 1235, 10.3390/ijms17081235.
- Mariangela Librizzi; Fabio CaraDonna; Ilenia Cruciata; Janusz Debski; Supojjanee Sansook; Michał Dadlez; John Spencer; Claudio Luparello; Molecular Signatures Associated with Treatment of Triple-Negative MDA-MB231 Breast Cancer Cells with Histone Deacetylase Inhibitors JAHA and SAHA. Chemical Research in Toxicology 2017, 30, 2187-2196, 10.1021/acs.chemrestox.7b00269.
- Claudio Luparello; Dalia Maria Lucia Asaro; Ilenia Cruciata; Storm Hassell-Hart; Supojjanee Sansook; John Spencer; Fabio CaraDonna; Cytotoxic Activity of the Histone Deacetylase 3-Selective Inhibitor Pojamide on MDA-MB-231 Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences 2019, 20, 804, 10.3390/ijms20040804.
- Meiling Huang; Jian Zhang; Changjiao Yan; Xiaohui Li; Juliang Zhang; Rui Ling; Small molecule HDAC inhibitors: Promising agents for breast cancer treatment. Bioorganic Chemistry 2019, 91, 103184, 10.1016/j.bioorg.2019.103184.
- Bumki Kim; An Overview of Naturally Occurring Histone Deacetylase Inhibitors. Current Topics in Medicinal Chemistry 2015, 14, 2759-2782, 10.2174/1568026615666141208105614.
- K C Lakshmaiah; Linu A Jacob; S Aparna; D Lokanatha; Smitha C Saldanha; Epigenetic therapy of cancer with histone deacetylase inhibitors.. Journal of Cancer Research and Therapeutics 2014, 10, 469-478, 10.4103/0973-1482.137937.
- Bin He; Longfei Dai; Xiaoqian Zhang; Diyu Chen; Jingbang Wu; Xiaode Feng; Yanpeng Zhang; Haiyang Xie; Lin Zhou; Jian Wu; et al.ShuSen Zheng The HDAC Inhibitor Quisinostat (JNJ-26481585) Supresses Hepatocellular Carcinoma alone and Synergistically in Combination with Sorafenib by G0/G1 phase arrest and Apoptosis induction. International Journal of Biological Sciences 2018, 14, 1845-1858, 10.7150/ijbs.27661.
- Igor Hrgovic; Monika Doll; Andreas Pinter; Roland Kaufmann; Stefan Kippenberger; Markus Meissner; Histone deacetylase inhibitors interfere with angiogenesis by decreasing endothelial VEGFR-2 protein half-life in part via a VE-cadherin-dependent mechanism. Experimental Dermatology 2017, 26, 194-201, 10.1111/exd.13159.
- Yanrong Su; Nathan R. Hopfinger; Theresa D. Nguyen; Thomas J. Pogash; Julia Santucci-Pereira; Jose Russo; Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. Journal of Experimental & Clinical Cancer Research 2018, 37, 1-18, 10.1186/s13046-018-0988-8.
- Fabiana As Brandão; Lorena S Derengowski; Patrícia Albuquerque; André M Nicola; Ildinete Silva-Pereira; Marcio J. Poças-Fonseca; Histone deacetylases inhibitors effects onCryptococcus neoformansmajor virulence phenotypes. Virulence 2015, 6, 618-630, 10.1080/21505594.2015.1038014.
- Vanita Chopra; Luisa Quinti; Prarthana Khanna; Paolo Paganetti; Rainer Kuhn; Anne B. Young; Aleksey G. Kazantsev; Steven M. Hersch; LBH589, A Hydroxamic Acid-Derived HDAC Inhibitor, is Neuroprotective in Mouse Models of Huntington’s Disease. Journal of Huntington's Disease 2016, 5, 347-355, 10.3233/jhd-160226.
- Elisabetta Soragni; Joel M. Gottesfeld; Translating HDAC inhibitors in Friedreich’s ataxia. Expert Opinion on Orphan Drugs 2016, 4, 961-970, 10.1080/21678707.2016.1215910.
- Jiun-I Lai; Luke J. Leman; Sherman Ku; Chris J. Vickers; Christian A. Olsen; Ana Montero; M. Reza Ghadiri; Joel M. Gottesfeld; Cyclic tetrapeptide HDAC inhibitors as potential therapeutics for spinal muscular atrophy: Screening with iPSC-derived neuronal cells. Bioorganic & Medicinal Chemistry Letters 2017, 27, 3289-3293, 10.1016/j.bmcl.2017.06.027.
- Margherita Brindisi; A. Prasanth Saraswati; Simone Brogi; Sandra Gemma; Stefania Butini; Giuseppe Campiani; Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. Journal of Medicinal Chemistry 2019, 63, 23-39, 10.1021/acs.jmedchem.9b00924.
- Mirella Vazzana; Monica Celi; Giulia Maricchiolo; Lucrezia Genovese; Valentina Corrias; Enza Maria Quinci; Giovanni De Vincenzi; Vincenzo Maccarrone; Gaetano Cammilleri; Salvatore Mazzola; et al.G. BuscainoFrancesco Filiciotto Are mussels able to distinguish underwater sounds? Assessment of the reactions of Mytilus galloprovincialis after exposure to lab-generated acoustic signals. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology 2016, 201, 61-70, 10.1016/j.cbpa.2016.06.029.
- Maria Giovanna Parisi; M. Mauro; G. Sarà; M. Cammarata; Temperature increases, hypoxia, and changes in food availability affect immunological biomarkers in the marine mussel Mytilus galloprovincialis. Journal of Comparative Physiology B 2017, 187, 1117-1126, 10.1007/s00360-017-1089-2.
- Mirella Vazzana; Maria Ceraulo; Manuela Mauro; Elena Papale; Maria Dioguardi; Salvatore Mazzola; Vincenzo Arizza; Marco Chiaramonte; Giuseppa Buscaino; Effects of acoustic stimulation on biochemical parameters in the digestive gland of Mediterranean mussel Mytilus galloprovincialis (Lamarck, 1819). The Journal of the Acoustical Society of America 2020, 147, 2414-2422, 10.1121/10.0001034.
- Mirella Vazzana; Manuela Mauro; Maria Ceraulo; Maria Dioguardi; Elena Papale; Salvatore Mazzola; Vincenzo Arizza; Francesco Beltrame; Luigi Inguglia; Giuseppa Buscaino; et al. Underwater high frequency noise: Biological responses in sea urchin Arbacia lixula (Linnaeus, 1758). Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology 2020, 242, 110650, 10.1016/j.cbpa.2020.110650.
- Mark E. Hay; Marine Chemical Ecology: Chemical Signals and Cues Structure Marine Populations, Communities, and Ecosystems. Annual Review of Marine Science 2009, 1, 193-212, 10.1146/annurev.marine.010908.163708.
- Lu Liu; Yao-Yao Zheng; Chang-Lun Shao; Chang-Yun Wang; Metabolites from marine invertebrates and their symbiotic microorganisms: molecular diversity discovery, mining, and application. Marine Life Science & Technology 2019, 1, 60-94, 10.1007/s42995-019-00021-2.
- Valentina Lazzara; Vincenzo Arizza; Claudio Luparello; Manuela Mauro; Mirella Vazzana; Bright Spots in The Darkness of Cancer: A Review of Starfishes-Derived Compounds and Their Anti-Tumor Action. Marine Drugs 2019, 17, 617, 10.3390/md17110617.
- Claudio Luparello; Debora Ragona; Dalia Maria Lucia Asaro; Valentina Lazzara; Federica Affranchi; Monica Celi; Vincenzo Arizza; Mirella Vazzana; Cytotoxic Potential of the Coelomic Fluid Extracted from the Sea Cucumber Holothuria tubulosa against Triple-Negative MDA-MB231 Breast Cancer Cells. Biology 2019, 8, 76, 10.3390/biology8040076.
- Claudio Luparello; Debora Ragona; Dalia Maria Lucia Asaro; Valentina Lazzara; Federica Affranchi; Vincenzo Arizza; Mirella Vazzana; Cell-Free Coelomic Fluid Extracts of the Sea Urchin Arbacia lixula Impair Mitochondrial Potential and Cell Cycle Distribution and Stimulate Reactive Oxygen Species Production and Autophagic Activity in Triple-Negative MDA-MB231 Breast Cancer Cells. Journal of Marine Science and Engineering 2020, 8, 261, 10.3390/jmse8040261.
- Claudio Luparello; Manuela Mauro; Valentina Lazzara; Mirella Vazzana; Collective Locomotion of Human Cells, Wound Healing and Their Control by Extracts and Isolated Compounds from Marine Invertebrates. Molecules 2020, 25, 2471, 10.3390/molecules25112471.
- Manuela Mauro; Valentina Lazzara; Diletta Punginelli; Vincenzo Arizza; Mirella Vazzana; Antitumoral compounds from vertebrate sister group: A review of Mediterranean ascidians. Developmental & Comparative Immunology 2020, 108, 103669, 10.1016/j.dci.2020.103669.
- De Laubenfels M.W.. The sponges of west-central Pacific; Oregon State Monographs: Corvallis OR USA, 1954; pp. 35-41.
- Doyeob Kim; Il Sun Lee; Jee Hyung Jung; Sung-Il Yang; Psammaplin A, a natural bromotyrosine derivative from a sponge, possesses the antibacterial activity against methicillin-resistantStaphylococcus aureus and the DNA gyrase-inhibitory activity. Archives of Pharmacal Research 1999, 22, 25-29, 10.1007/bf02976431.
- J.N Tabudravu; V.G.H Eijsink; G.W Gooday; Marcel Jaspars; D Komander; M Legg; B Synstad; D.M.F Van Aalten; Psammaplin A, a chitinase inhibitor isolated from the fijian marine sponge Aplysinella rhax. Bioorganic & Medicinal Chemistry 2002, 10, 1123-1128, 10.1016/s0968-0896(01)00372-8.
- Yahong Jiang; Eun-Young Ahn; Seung Hee Ryu; Dong-Kyoo Kim; Jang-Su Park; Hyun Joo Yoon; Song You; Burm-Jong Lee; Seung Hoon Lee; Jee H. Jung; et al. Cytotoxicity of psammaplin A from a two-sponge association may correlate with the inhibition of DNA replication. BMC Cancer 2004, 4, 70-70, 10.1186/1471-2407-4-70.
- Dong Hoon Kim; Jongheon Shin; Ho Jeong Kwon; Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Experimental & Molecular Medicine 2007, 39, 47-55, 10.1038/emm.2007.6.
- Ivette C. Piña; Jeffrey T. Gautschi; Gui-Yang-Sheng Wang; Miranda L. Sanders; Francis J. Schmitz; Dennis France; Susan Cornell-Kennon; Lidia C. Sambucetti; Stacy W. Remiszewski; Larry B. Perez; et al.Kenneth W. BairPhillip Crews Psammaplins from the SpongePseudoceratinapurpurea:Inhibition of Both Histone Deacetylase and DNA Methyltransferase. The Journal of Organic Chemistry 2003, 68, 3866-3873, 10.1021/jo034248t.
- Flor D. Mora; Deborah K. Jones; Prashant V. Desai; Akshay Patny; Mitchell A. Avery; Dennis R. Feller; Troy Smillie; Yu-Dong Zhou; Dale G. Nagle; Bioassay for the Identification of Natural Product-Based Activators of Peroxisome Proliferator-Activated Receptor-γ (PPARγ): The Marine Sponge Metabolite Psammaplin A Activates PPARγ and Induces Apoptosis in Human Breast Tumor Cells. Journal of Natural Products 2006, 69, 547-552, 10.1021/np050397q.
- Matthias G. J. Baud; Thomas Leiser; Patricia Haus; Sharon Samlal; Ai Ching Wong; Robert J. Wood; Vanessa Petrucci; Mekala Gunaratnam; Siobhan M. Hughes; Lakjaya Buluwela; et al.Fabrice TurlaisStephen NeidleFranz-Josef Meyer-AlmesAndrew J. P. WhiteMatthew J. Fuchter Defining the Mechanism of Action and Enzymatic Selectivity of Psammaplin A against Its Epigenetic Targets. Journal of Medicinal Chemistry 2012, 55, 1731-1750, 10.1021/jm2016182.
- Yu-Dong Zhou; Jun Li; Lin Du; Fakhri Mahdi; Thuy P. Le; Wei-Lun Chen; Steve M. Swanson; Kounosuke Watabe; Dale G. Nagle; Biochemical and Anti-Triple Negative Metastatic Breast Tumor Cell Properties of Psammaplins. Marine Drugs 2018, 16, 442, 10.3390/md16110442.
- Mee Young Ahn; Jee H. Jung; Yong Jin Na; Hyung Sik Kim; A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecologic Oncology 2008, 108, 27-33, 10.1016/j.ygyno.2007.08.098.
- Prashant Mujumdar; Kanae Teruya; Kathryn F. Tonissen; Daniela Vullo; Claudiu T. Supuran; Thomas S. Peat; Sally-Ann Poulsen; An Unusual Natural Product Primary Sulfonamide: Synthesis, Carbonic Anhydrase Inhibition, and Protein X-ray Structures of Psammaplin C. Journal of Medicinal Chemistry 2016, 59, 5462-5470, 10.1021/acs.jmedchem.6b00443.
- Iris Chiara Salaroglio; Prashant Mujumdar; Laura Annovazzi; Joanna Kopecka; Marta Mellai; Davide Schiffer; Sally-Ann Poulsen; Chiara Riganti; Carbonic Anhydrase XII Inhibitors Overcome P-Glycoprotein–Mediated Resistance to Temozolomide in Glioblastoma. Molecular Cancer Therapeutics 2018, 17, 2598-2609, 10.1158/1535-7163.mct-18-0533.
- Prashant Mujumdar; Joanna Kopecka; Silvia Bua; Claudiu T. Supuran; Chiara Riganti; Sally-Ann Poulsen; Carbonic Anhydrase XII Inhibitors Overcome Temozolomide Resistance in Glioblastoma. Journal of Medicinal Chemistry 2019, 62, 4174-4192, 10.1021/acs.jmedchem.9b00282.
- Edward A. Ratovitski; Tumor Protein (TP)-p53 Members as Regulators of Autophagy in Tumor Cells upon Marine Drug Exposure. Marine Drugs 2016, 14, 154, 10.3390/md14080154.
- Woong Sub Byun; Won Kyung Kim; Hae Ju Han; Hwa-Jin Chung; Kyungkuk Jang; Han Sun Kim; Sunghwa Kim; Donghwa Kim; Eun Seo Bae; Sunghyouk Park; et al.Jeeyeon LeeHyeung-Geun ParkSang Kook Lee Targeting Histone Methyltransferase DOT1L by a Novel Psammaplin A Analog Inhibits Growth and Metastasis of Triple-Negative Breast Cancer. Molecular Therapy - Oncolytics 2019, 15, 140-152, 10.1016/j.omto.2019.09.005.
- Kentaro Takada; Yasufumi Imae; Yuji Ise; Susumu Ohtsuka; Akihiro Ito; Shigeru Okada; Minoru Yoshida; Shigeki Matsunaga; Yakushinamides, Polyoxygenated Fatty Acid Amides That Inhibit HDACs and SIRTs, from the Marine Sponge Theonella swinhoei. Journal of Natural Products 2016, 79, 2384-2390, 10.1021/acs.jnatprod.6b00588.
- Fumiaki Nakamura; Norio Kudo; Yuki Tomachi; Akiko Nakata; Misao Takemoto; Akihiro Ito; Hodaka Tabei; Daisuke Arai; Nicole De Voogd; Minoru Yoshida; et al.Yoichi NakaoNobuhiro Fusetani Halistanol sulfates I and J, new SIRT1–3 inhibitory steroid sulfates from a marine sponge of the genus Halichondria. The Journal of Antibiotics 2017, 71, 273-278, 10.1038/ja.2017.145.
- Yoichi Nakao; Satoru Yoshida; Shigeki Matsunaga; Nobuaki Shindoh; Yoh Terada; Koji Nagai; Jun K. Yamashita; A. Ganesan; Rob W. M. Van Soest; Nobuhiro Fusetani; et al. Azumamides A–E: Histone Deacetylase Inhibitory Cyclic Tetrapeptides from the Marine SpongeMycale izuensis. Angewandte Chemie International Edition 2006, 45, 7553-7557, 10.1002/anie.200602047.
- Shijun Wen; Krystle L. Carey; Yoichi Nakao; Nobuhiro Fusetani; Graham Packham; A. Ganesan; Total Synthesis of Azumamide A and Azumamide E, Evaluation as Histone Deacetylase Inhibitors, and Design of a More Potent Analogue. Organic Letters 2007, 9, 1105-1108, 10.1021/ol070046y.
- Nakia Maulucci; Maria Giovanna Chini; Simone Di Micco; Irene Izzo; Emiddio Cafaro; Adele Russo; Paola Gallinari; Chantal Paolini; Maria Chiara Nardi; Agostino Casapullo; et al.Raffaele RiccioGiuseppe BifulcoFrancesco De Riccardis Molecular Insights into Azumamide E Histone Deacetylases Inhibitory Activity. Journal of the American Chemical Society 2007, 129, 3007-3012, 10.1021/ja0686256.
- Yoichi Nakao; Genta Narazaki; Takuhiro Hoshino; Satoko Maeda; Minoru Yoshida; Hiroshi Maejima; Jun K. Yamashita; Evaluation of antiangiogenic activity of azumamides by the in vitro vascular organization model using mouse induced pluripotent stem (iPS) cells. Bioorganic & Medicinal Chemistry Letters 2008, 18, 2982-2984, 10.1016/j.bmcl.2008.03.053.
- Jesper S. Villadsen; Helle M. Stephansen; Alex R. Maolanon; Pernille Harris; Christian A. Olsen; Total Synthesis and Full Histone Deacetylase Inhibitory Profiling of Azumamides A–E as Well as β2- epi-Azumamide E and β3-epi-Azumamide E. Journal of Medicinal Chemistry 2013, 56, 6512-6520, 10.1021/jm4008449.
- Alex R. Maolanon; Jesper S. Villadsen; Niels Johan Christensen; Casper Hoeck; Tina Friis; Pernille Harris; Charlotte Held Gotfredsen; Peter Fristrup; Christian A. Olsen; Methyl Effect in Azumamides Provides Insight Into Histone Deacetylase Inhibition by Macrocycles. Journal of Medicinal Chemistry 2014, 57, 9644-9657, 10.1021/jm501399d.
- Naoya Oku; Koji Nagai; Nobuaki Shindoh; Yoh Terada; Rob W.M. Van Soest; Shigeki Matsunaga; Nobuhiro Fusetani; Three new cyclostellettamines, which inhibit histone deacetylase, from a marine sponge of the genus Xestospongia. Bioorganic & Medicinal Chemistry Letters 2004, 14, 2617-2620, 10.1016/j.bmcl.2004.02.062.
- Yoonyeong Lee; Kyoung Hwa Jang; Ju-Eun Jeon; Woo-Young Yang; Chung Ja Sim; Ki-Bong Oh; Jongheon Shin; Kyung Hwa Jang; Cyclic Bis-1,3-dialkylpyridiniums from the Sponge Haliclona sp.. Marine Drugs 2012, 10, 2126-2137, 10.3390/md10092126.
- Shou-Ping Shih; Man-Gang Lee; Mohamed El-Shazly; Yung-Shun Juan; Zhi-Hong Wen; Ying-Chi Du; Jui-Hsin Su; Ping-Jyun Sung; Yu-Cheng Chen; Juan-Cheng Yang; et al.Yang-Chang WuMei-Chin Lu Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Marine Drugs 2015, 13, 3132-3153, 10.3390/md13053132.
- Motoki Takaku; Takashi Kainuma; Takako Ishida-Takaku; Shintaro Ishigami; Hidekazu Suzuki; Satoshi Tashiro; Rob W. M. Van Soest; Yoichi Nakao; Hitoshi Kurumizaka; Halenaquinone, a chemical compound that specifically inhibits the secondary DNA binding of RAD51. Genes to Cells 2011, 16, 427-436, 10.1111/j.1365-2443.2011.01494.x.
- Sachiko Tsukamoto; Tomoharu Takeuchi; Tetsuro Kawabata; Hikaru Kato; Michiko Yamakuma; Kanae Matsuo; Ahmed H. El-Desoky; Fitje Losung; Remy E.P. Mangindaan; Nicole J. De Voogd; et al.Yoichiro ArataHideyoshi Yokosawa Halenaquinone inhibits RANKL-induced osteoclastogenesis. Bioorganic & Medicinal Chemistry Letters 2014, 24, 5315-5317, 10.1016/j.bmcl.2014.09.043.
- Eric H. Andrianasolo; Liti Haramaty; Eileen White; Richard A. Lutz; Paul G. Falkowski; Mode of Action of Diterpene and Characterization of Related Metabolites from the Soft Coral, Xenia elongata. Marine Drugs 2014, 12, 1102-1115, 10.3390/md12021102.
- Hilaire V. Kemami Wangun; Alexander Wood; Catherine Fiorilla; John K. Reed; Peter J. McCarthy; Amy E. Wright; Gymnochromes E and F, Cytotoxic Phenanthroperylenequinones from a Deep-Water Crinoid,Holopus rangii. Journal of Natural Products 2010, 73, 712-715, 10.1021/np900526y.
- Anthony R. Carroll; Brent R. Copp; Rohan A. Davis; Robert A. Keyzers; Michele Prinsep; Marine natural products. Natural Product Reports 2020, 37, 175-223, 10.1039/c9np00069k.
- Shang Li; Hequan Yao; Jinyi Xu; Sheng Jiang; Synthetic Routes and Biological Evaluation of Largazole and Its Analogues as Potent Histone Deacetylase Inhibitors. Molecules 2011, 16, 4681-4694, 10.3390/molecules16064681.
- T.A. Varghese; M.A. Jayasri; Suthindhiran K; Marine Actinomycetes as potential source for histone deacetylase inhibitors and epigenetic modulation. Letters in Applied Microbiology 2015, 61, 69-76, 10.1111/lam.12430.
- Wael Houssen; Marcel Jaspars; 4-Hydroxybenzoyl Derivative from the Aqueous Extract of the HydroidCampanulariasp.. Journal of Natural Products 2005, 68, 453-455, 10.1021/np049666n.



