 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yongming Sang | + 1793 word(s) | 1793 | 2020-11-30 07:54:19 | | | |
| 2 | Catherine Yang | Meta information modification | 1793 | 2020-12-03 04:24:03 | | |
Video Upload Options
SARS-CoV2 has caused the current pandemic of new coronavirus disease 2019 (COVID-19) worldwide. Clinical outcomes of COVID-19 illness range broadly from asymptotic and mild to a life-threatening situation. This casts uncertainties for defining host determinants underlying the disease severity. Recent genetic analyses based on extensive clinical sample cohorts using genome-wide association studies (GWAS) and high throughput sequencing curation revealed genetic errors and gene loci associated with about 20% of life-threatening COVID-19 cases. Significantly, most of these critical genetic loci are enriched in two immune signaling pathways, i.e., interferon-mediated antiviral signaling and chemokine-mediated/inflammatory signaling. In line with these genetic profiling studies, the broad spectrum of COVID-19 illness could be explained by immuno-pathological regulation of these critical immunogenetic pathways through various epigenetic mechanisms, which further interconnect to other vital components such as those in the renin–angiotensin–aldosterone system (RAAS) because of its direct interaction with the virus causing COVID-19. Together, key genes unraveled by genetic profiling may provide targets for precisely early risk diagnosis and prophylactic design to relieve severe COVID-19. The confounding epigenetic mechanisms may be key to understanding the clinical broadness of COVID-19 illness.
1. The Broad Spectrum and Critical Illness in COVID-19 Progression
The coronavirus disease 2019 (COVID-19), which has been declared a worldwide pandemic by the WHO since March of 2020, is caused by the novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1][2][3][4]. The virus evolves at a highly contagious rate in human beings, with a basic reproduction number (R0) ranging at 1.4–5.7. The clinical outcome of COVID-19 varies broadly among infected people, ranging from asymptotic infection and common cold-like sickness to a severe pneumonia leading to acute respiratory distress syndrome (ARS) and multi-organ complications that potentially have fatal prognosis [5][6][7][8]. Complications of severe COVID-19 include vasculitis, coagulopathy, thrombosis, septic shock, and even multi-organ failure [5][6][7][8]. The epidemiology of COVID-19 shows a diverse pattern across people who are different in age, sex, ethnicity, and particularly among those with pre-existing medical conditions [6][7][8][9][10][11]. For example, the US statistics showed that older patients (aged ≥65 years) accounted for 31% of all cases, 45% of hospitalizations, 53% admissions of intensive care unit (ICU), and 80% of deaths, with the highest incidence of severe outcomes in patients aged ≥85 years [1][4][8]. Similarly, increased risk of critical and life-threatening illnesses was reported to associate with males and particularly pre-existing comorbidities, including cardiovascular, renal, liver, diabetes, and other autoimmune diseases as well as obesity condition [4][5][6][7][8][9][10][11]. In contrast, evidence indicates that children (median age 4–7 years) have a lower susceptibility and risk for critical illness. However, under the circumstance of comorbidity and genetic risks, the disparity of the risk for severe COVID-19 becomes vague concerning the factors of age, sex, and ethnicity [4][5][6][7][8][9][10][11]. With a critical viral disease like COVID-19, illness comes from both the virus infection and interacting with immune responses, especially a consequential imbalance of harmful immunopathies over proper immune responses. Upon exposure to the same virus, whereas individuals show asymptotic or mild illness plausibly mounting effective immune reactions, severe COVID-19 patients, however, may reflect dysfunctional immune reactions that further leads to pathological exacerbation accompanying uncontrolled virus spreading and immune overwhelming [9][10][11][12][13][14][15][16][17]. As the virological branch focuses on diminishing viral spreading and virulence to cause disease, deciphering the genetic and especially epigenetic associations underlie severe COVID-19 will grasp the immunogenetic theme for severity prognosis in the host, thus providing manageable targets for early risk diagnosis and development of prophylactic and therapeutic remedies to face current pandemic [18][19][20].
2. Genetic Association: Interferon and Chemokine Response Representing the Centric Immune Determinants Underlying Severe COVID-19
About two months post the WHO declaring the COVID-19 pandemic, a global initiative of COVID-19 host genetics was commenced to elucidate the role of host genetic factors in SARS-CoV2 susceptibility and COVID-19 severity [21]. The first report about genome-wide association study (GWAS) of severe COVID-19 with ARS detected two genetic susceptibility loci at Chr3p21.31 and Chr9q34.2 using a meta-analysis of the two case/control panels including 835/1225 and 775/950 samples from Italy and Spain, respectively (Table 1) [22]. Significantly, the association within the locus Chr3p21.31 spans the genes SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, XCR1, CCR1, and CCR3 (gene symbols are standard ones from NCBI, see Figure 1 legend for definitions of abbreviations), which include several chemokine receptors (CCRs, CXCR6, and XCR1) mediating chemokine signaling pathways for leukocyte chemotaxis, inflammatory regulation and relevant immunopathies causing lung injury. Notably, Chr3p21.31 locus has been reproducibly associated with severe COVID-19 by at least three GWAS studies, indicating it constitutes a common genetic mechanism underlying severe COVID-19 [22][23][24]. Interestingly, an independent study also identified the ~50 kb region of locus Chr3p21.31 representing an allelic risk that was inherited from Neanderthals and is carried by ~50% of people in South Asia and ~16% of people in Europe today, who were predicted to be prone to the progression of severe COVID-19 [24]. In addition, the association of Chr3p21.31 locus was also reflected by the critical illness in the younger patients (<65 years) with less comorbidity, indicating a de facto genetic correlation [22][23][24]. Several clinical observations correlated blood types with the severity of COVID-19, i.e., O blood type seems more protective compared to a higher risk of non-O, especially A blood type [25][26][27]. One GWAS assay using two case-control European panels associated severe-COVID-19 with locus Chr9q34.2, which concurs the ABO blood group locus [22]. However, this association was not significantly demonstrated in two other GWAS assays published [23][24], indicating that the association of blood type locus with severe COVID-19 is not as universal as the chemokine receptor locus at Chr3p21.31 (Table 1). Therefore, more studies are needed to extensively verify the association of blood types with the progression of COVID-19 severity. In addition, no mechanistic research about the association of blood types with COVID-19 severity has been reported. In general, antigens determining blood types can serve as direct receptors or co-factors for some pathogenic infections; indirectly, many blood group antigens facilitate cell adhesion, substance intake and signaling transduction [22][25][26][27]. Given the reported inconsistency on association of blood types with COVID-19 severity, we interpret an indirect role (such as regulation through the RAAS system, see next section) of blood types on COVID-19 susceptibility and disease progression [22][25][26][27].
Pairo-Castineira et al. released their GWAS analysis using a bigger case/control cohort (2244/10220) from UK hospitals, which represent >95% of all ICU beds in the UK (Table 1) [23]. In addition to the detection of a strong association signal at the Chr3p21.31 locus, the study identified and replicated four novel genome-wide significant associations. These include: (1) at Chr6p22.1–33 region spanning major histocompatibility complex, class I-G, HLA-G, and Coiled-Coil Alpha-Helical Rod Protein 1, CCHCR1 genes; (2) at Chr19p13.3 locus within the gene encoding dipeptidyl peptidase 9 (DPP9); (3) at Chr12q24.13 locus spanning a gene cluster encoding antiviral restriction enzyme activators (OAS1, OAS2, OAS3); and (4) at Chr21q22.1 spanning the interferon receptor gene IFNAR2) [23]. Elegantly, the study also supplemented GWAS illumination with evidence using Mendelian randomization (MR) and transcriptome-wide association (TWAS) assays to define a causal link from the low expression of IFNAR2, and high expression of TYK2, to life-threatening COVID-19. TWAS in lung tissue determined the association of severe COVID-19 with increased expression of the monocyte/macrophage chemotactic receptor CCR2 [23]. Collectively, this study robustly determined genetic signals relating to key host antiviral defense mechanisms, especially that mediated by interferon (IFN)-signaling and chemokine receptors in orchestrating chemotactic and inflammatory responses as clinically demonstrated commonly in severe Covid-19 cases (Figure 1) [12][13][14][28][29][30].
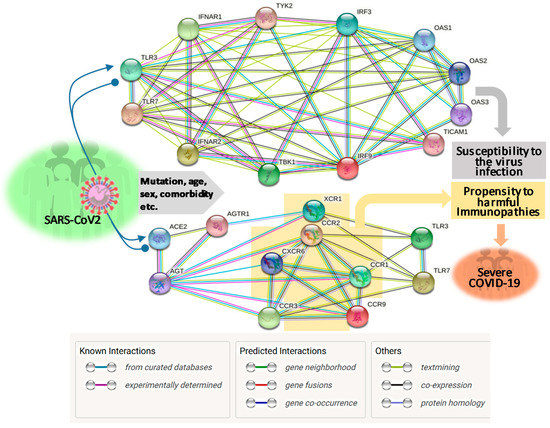
Using an approach combining both next-generation sequencing (NGS) and experimental validation, Zhang et al. elucidated an enrichment of genetic risk variants at thirteen human loci governing the Toll-like receptor (TLR)-3- and IFN-regulatory factor (IRF)-7-dependent type I IFN immunity in 659 patients with life-threatening COVID-19 [31]. In contrast, few of these genetic risk variants were detected in the 534 control subjects with asymptomatic or benign infection. These 13 genetic risk loci displayed functional deficiency of these immune genes. They accounted for 3.5% of severe COVID-19 patients aged 17 through 77 years, and progressed to a life-threatening pneumonia without prior severe infection, indicating a determining role of dysfunctional IFN-mediated antiviral immunity underlying the progression of severe COVID-19 (Table 1 and Figure 1) [31].
Table 1. Genetic inborn or epigenetic obtaining errors associated with severe COVID-19 *.
| Chr. Location (Key Genes Covered, or Epigenetic Effect) |
Association (Appr./OR: Freq.) |
Major Immune Pathway Involved |
References & Notes |
|---|---|---|---|
| 3p21.31 (SLC6A20, LZTFL1, FYCO1, CXCR6, XCR1, and CCR9; Neanderthal-originated allelic region) |
GWAS 95% CI/ (1.95–2.79: 1610 vs. 2205) [22] (2.14: 2244 vs. ~5X 2244) [23] |
ACE2 mediated amino acid transport (SLC6A20); Chemokine and Inflammation signaling, chemotaxis, immunopathies for lung injury (others) |
Associated at [22][23][24] |
| 6p22.1–33 (HLA-G, CCHCR1, NOTCH4) |
GWAS 95% CI/ (1.30–1.85: 2244 vs. ~5X 2244) |
Antigen processing and presentation (HLA); P-body component for RNA metabolism, associated with psoriasis (CCHCR1); lymphocyte development (NOTCH4) | Associated by [23] |
| 9q34.2 (ABO blood type locus) |
GWAS 95% CI/ (1.37–1.45: 1610 patient vs. 2205 control) |
Blood type-dependent pathological reaction, such as coagulation and thrombolysis | Associated by [22] |
| 12q24.13 (OAS1, OAS2, OAS3) |
GWAS 95% CI/ (1.29: 2244 vs. ~5X 2244) [23] |
IFN-mediated antiviral signaling | Associated by [23] |
| 19p13.3 (DPP9, TYK2) |
GWAS 95% CI/ (1.36–1.59: 2244 vs. ~5X 2244) [23] |
Innate antiviral defense (TYK2), and antigen presentation, CXCL10 signaling, and associated to obesity, diabetes, and cancer (DPP9) | Associated by [23] |
| 21q22.1 (IFNAR2) |
GWAS 95% CI/ (1.28: 2244 vs. ~5X 2244) [23] |
IFN-mediated immune signaling | Associated by [23] |
| Several Chr. (TLR3, UNC93B1, TICAM1, TRAF3, TBK1, IRF3/7/9, IFNAR1/2, STAT1/2) |
NGS and variant calling, wet-bench validation (3.5% of 659 severe COVID-19 vs few in 534 control) |
IFN mediated immune signaling |
Detected by [31] |
| Epigenetic obtaining (Autoantibody against IFNs, 94% in male) |
Wet-bench detection (13.7% of 987 severe COVID-19 vs. 0.33% in 1227 control) |
IFN mediated immune signaling | Detected by [32] |
| Epigenetic obtaining (Higher incidence of severe COVID-19 in aged, male, and comorbid patients) |
Inclusive studies and evidence (Higher incidence of severe COVID-19 in aged, male, and comorbid patients) |
Dysregulated IFN and chemokine responses, chronic/systemic inflammation, impaired other immune responses | Exemplified by [18][19][20][32][33][34] |
* Defined as accompanying respiratory failure in hospitalized patients. Abbreviation: Appr., approaches to associate the gene loci with severe COVID-19 in the references; CI, confidence intervals; Chr., human chromosome from genome build hg38; GWAS, genome wide association study; IGV, integrative genomics viewer; NGS, next-generation sequencing; OR: Freq., odds ratio: frequency in severe COVID-19 patient vs in the control groups. The gene symbols are standard ones from NCBI, see Figure 1 legend for definitions.
References
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 25 October 2020).
- Epidemiology Working Group for NCIP Epidemic Response, Chinese Center for Disease Control and Prevention. The Epidemiological Characteristics of an Outbreak of 2019 Novel Coronavirus Diseases (COVID-19). Zhonghua Liu Xing Bing Xue Za Zhi 2020, 41, 145–151.
- World Health Organization. Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19) [Pdf]. Available online: https://www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report.pdf (accessed on 25 October 2020).
- COVID-19 Pandemic Planning Scenarios. Available online: https://www.cdc.gov/coronavirus/2019-ncov/hcp/planning-scenarios.html (accessed on 25 October 2020).
- Coronavirus disease 2019 (COVID-19). Available online: https://bestpractice.bmj.com/topics/en-gb/3000201 (accessed on 25 October 2020).
- Sanche, S.; Lin, Y.T.; Xu, C.; Romero-Severson, E.; Hengartner, N.; Ke, R. High Contagiousness and Rapid Spread of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1470–1477.
- Courtney, E.P.; Goldenberg, J.L.; Boyd, P. The contagion of mortality: A terror management health model for pandemics. Br. J. Soc. Psychol. 2020, 59, 607–617.
- Age, Sex, Existing Conditions of COVID-19 Cases and Deaths. Available online: https://www.worldometers.info/coronavirus/coronavirus-age-sex-demographics/ (accessed on 25 October 2020).
- Scully, E.P.; Haverfield, J.; Ursin, R.L.; Tannenbaum, C.; Klein, S.L. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat. Rev. Immunol. 2020, 20, 442–447.
- Gebhard, C.; Regitz-Zagrosek, V.; Neuhauser, H.K.; Morgan, R.; Klein, S.L. Impact of sex and gender on COVID-19 outcomes in Europe. Biol. Sex. Differ. 2020, 11, 29.
- Jutzeler, C.R.; Bourguignon, L.; Weis, C.V.; Tong, B.; Wong, C.; Rieck, B.; Pargger, H.; Tschudin-Sutter, S.; Egli, A.; Borgwardt, K.; et al. Comorbidities, clinical signs and symptoms, laboratory findings, imaging features, treatment strategies, and outcomes in adult and pediatric patients with COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 37, 101825.
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; Breillat, P.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.
- Acharya, D.; Liu, G.; Gack, M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 397–398.
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Morieira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941.
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.
- Schultheiß, C.; Paschold, L.; Simnica, D.; Mohme, M.; Willscher, E.; von Wenserski, L.; Scholz, R.; Wieters, I.; Dahlke, C.; Tolosa, E.; et al. Next-Generation Sequencing of T and B Cell Receptor Repertoires from COVID-19 Patients Showed Signatures Associated with Severity of Disease. Immunity 2020, 53, 442–455.
- Mantovani, A.; Netea, M.G. Trained Innate Immunity, Epigenetics, and Covid-19. N. Engl. J. Med. 2020, 383, 1078–1080.
- Kleen, T.O.; Galdon, A.A.; MacDonald, A.S.; Dalgleish, A.G. Mitigating Coronavirus Induced Dysfunctional Immunity for At-Risk Populations in COVID-19: Trained Immunity, BCG and “New Old Friends”. Front. Immunol. 2020, 11, 2059.
- Sang, E.R.; Tian, Y.; Miller, L.C.; Sang, Y. Epigenetic Evolution of ACE2 and IL-6 Genes as Non-Canonical Interferon-Stimulated Genes Correlate to COVID-19 Susceptibility in Vertebrates. bioRxiv 2020.
- COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020, 28, 715–718.
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534.
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.; Rawlik, K.; Parkinson, N.; Pasko, D.; Walker, S.; Richmond, A.; Fourman, M.H.; et al. Genetic mechanisms of critical illness in Covid-19. medRxiv 2020.
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020.
- Latz, C.A.; DeCarlo, C.; Boitano, L.; Png, C.Y.M.; Patell, R.; Conrad, M.F.; Eagleton, M.; Dua, A. Blood type and outcomes in patients with COVID-19. Ann. Hematol. 2020, 99, 2113–2118.
- Gérard, C.; Maggipinto, G.; Minon, J.M. COVID-19 and ABO blood group: Another viewpoint. Br. J. Haematol. 2020, 190, e93–e94.
- Wu, Y.; Feng, Z.; Li, P.; Yu, Q. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin. Chim. Acta 2020, 509, 220–223.
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. Covid-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582.
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702.
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabil, I.K.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570.
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585.
- Chlamydas, S.; Papavassiliou, A.G.; Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 2020, 30, 1–8.
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410.


