 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesca Becherucci | + 2002 word(s) | 2002 | 2020-11-20 10:41:45 | | | |
| 2 | Vicky Zhou | + 104 word(s) | 2106 | 2020-11-26 03:21:55 | | |
Video Upload Options
The genetic landscape of steroid-resistant nephrotic syndrome (SRNS) is dramatically changing. Extended genetic testing is a key tool in understanding the pathophysiology of the disease and in identifying a precise etiology, with important implications in the clinical management of patients. However, forward genetics (i.e. moving from phenotype to genotype) is frequently insufficient. In this view, advancing and finishing diagnostic strategies, including reverse genetics and phenocopies identification, is pivotal in improving the approach to patients with SRNS.
1. Introduction
Nephrotic syndrome (NS) is a clinical picture common to different glomerular and non-glomerular diseases, with variable response to treatments and heterogeneous outcomes. In children and adolescents, primary NS is classified as steroid-sensitive (SSNS) and steroid-resistant NS (SRNS) based on the response to standard steroid treatment. Patients with SRNS have a significantly worse prognosis due to an increased risk of developing chronic kidney disease (CKD) and severe side effects of immunosuppressive therapies that are commonly used as second-line treatments.
Genetic testing has become a valuable diagnostic tool in defining the etiology of SRNS, leading to the identification of a genetic cause in about 30% of patients (monogenic podocytopathies). Genetic SRNS has no chance to respond to immunosuppressive therapies and frequently progresses to end-stage kidney disease (ESKD), accounting for about 15% of pediatric cases [1][2]. The identification of genetic SRNS is pivotal in order to tailor therapeutic measures and prognosis prediction, as well as genetic counseling for reproductive and organ transplantation purposes.
The advent and spreading of next-generation sequencing (NGS) have significantly improved diagnostic strategies for inherited diseases, including SRNS. Indeed, simultaneous sequencing of multiple genes has become feasible, time- and cost-effective and is increasingly performed, either in diagnostics or in research. However, after an initial phase, widening the number of genes analyzed by NGS in patients with SRNS did not result in a significant improvement in the diagnostic rate of genetic forms that stabilized around 30%, thus hindering the advent of precision medicine in SRNS. Recent evidence supports the hypothesis that “phenocopies” could account for a non-negligible fraction of SRNS patients who are currently classified as non-genetic upon standard genetic testing (i.e., genetic analysis limited to monogenic podocytopathies) [3][4], paving the way for a more comprehensive understanding of the overall genetic background of each single patient and for the elucidation of potential genetic biomarkers of disease progression in SRNS. Following its first introduction in medicine, the term phenocopies is currently used to identify patients sharing the same phenotype who are carriers of different genotypes. Genetic testing is crucial for detecting phenocopies. However, it can be insufficient.
2. From Phenotype to Genotype and Backwards in SRNS
Thanks to the improvements and cost-cut of massive parallel sequencing techniques, in recent years, several studies have been carried out using NGS in large cohorts of patients affected by kidney diseases, paving the way for its use in the standard clinical work-up, when genetic etiology is suspected. Genetic testing allows clinicians to test the hypothesis that a phenotype is caused by mutations in disease-causing genes (moving from phenotype to genotype, Figure 1A). The continuous growth in the number of genes reported in association with a specific disease (phenotype) makes NGS ideal to address this diagnostic issue (genotype). However, the spreading use of NGS forces geneticists and clinicians to deal with the problem of unexpected results (i.e., mutations in genes not previously reported in association with the phenotype) and with the challenge of disease reclassification, especially with whole-exome sequencing (WES; i.e. sequencing of all coding region of DNA). WES has proven to be useful for the reclassification of various inherited nephropathies, such as nephronophthisis, ciliopathies and CAKUT [5][6][7][8][9][10][11][12][13]. Strikingly, in recent years, WES has been successfully applied also in unselected cohorts of adult patients with ESKD of unknown origin, identifying a monogenic cause in nearly 10% of cases who were therefore reclassified [14][15]. As above mentioned, patients presenting with a phenotype corresponding to a specific hereditary disease but not carrying the expected genotype can be considered phenocopies. However, establishing a causative relationship between (unexpected) genotype and phenotype (moving from genotype to phenotype, also known as reverse genetics) could be tiresome. Very recently, the genetics of SRNS started this challenge.
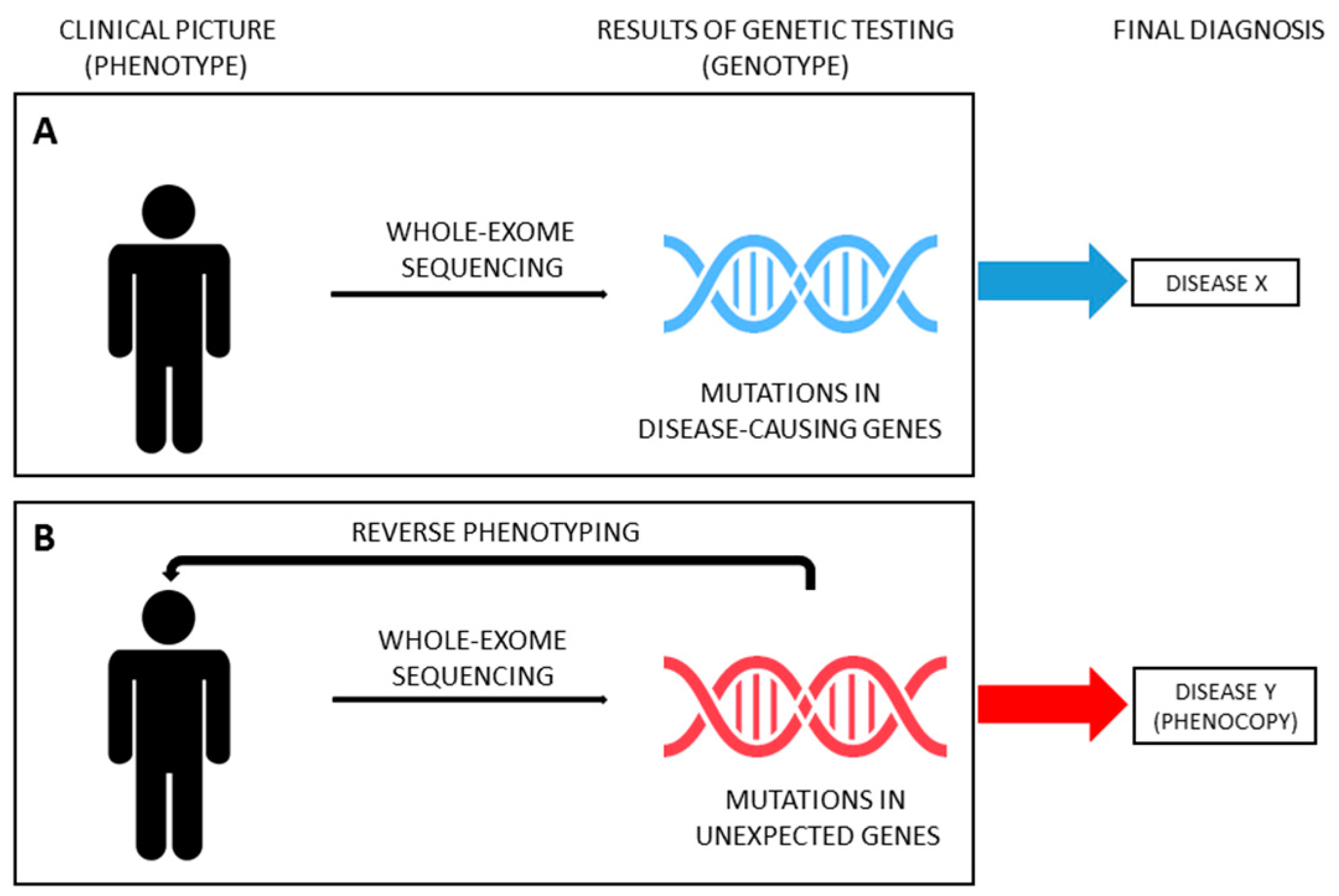
Genetic testing with NGS has become a reliable and affordable diagnostic tool in patients with early-onset idiopathic SRNS, leading to an improvement in the personalized management of about 30% of cases who result as being affected by a genetic disorder caused by mutations in one of about 30 genes responsible for monogenic podocytopathies [16][17]. However, a genetic etiology has been claimed for many SRNS patients classified as non-genetic by currently performed genetic testing.
Despite being usually recognized by standard diagnostic work-up because of clinical features suggestive of the syndromic picture, genetic nephropathies outside the podocytopathies spectrum (e.g., Alport syndrome, Dent disease) can rarely present as isolated SRNS or focal segmental glomerulosclerosis (FSGS), preventing a precise diagnosis and leading to misclassification [18][19][20][21][22][23][24]. Indeed, genetic testing for podocytopathy genes is negative. According to the general concept, these conditions can be named phenocopies of monogenic podocytopathies, since they present with a clinical picture of apparently isolated SRNS but they are not caused by mutations in podocytes genes [25][26][27][28][29]. A comprehensive genetic testing in SRNS should therefore comprise phenocopy genes in order to provide a conclusive diagnosis and correctly classify disease entities (Table 1).
Table 1. Phenocopy genes. This is a table illustrating phenocopy genes and the disease usually caused by their mutations (phenotype).
| Gene | Phenotype | Ref. | |
|---|---|---|---|
| CLCN5 | CHLORIDE CHANNEL 5 | Dent disease | [3][4][20][21][30] |
| COL4A3 | COLLAGEN, TYPE IV, ALPHA-3 | Alport syndrome | [3][4][14][25][31][32][33] |
| COL4A4 | COLLAGEN, TYPE IV, ALPHA-4 | Alport syndrome | [14][18][25][32] |
| COL4A5 | COLLAGEN, TYPE IV, ALPHA-5 | Alport syndrome | [3][14][32][33] |
| GLA | GALACTOSIDASE, ALPHA | Fabry disease | [3][4][33] |
| AGXT | ALANINE-GLYOXYLATE AMINOTRANSFERASE | Hyperoxaluria, primary, type 1 | [3] |
| CTNS | CYSTINOSIN | Cystinosis | [3][4] |
| FN1 | FIBRONECTIN 1 | Glomerulopathy with fibronectin deposits 2 | [3] |
| WDR19 | WD REPEAT-CONTAINING PROTEIN 19 | Nephronophthisis 13 | [3] |
| LAMB2 | LAMININ, BETA-2 | Pierson syndrome | [4] |
| FAT1 | FAT ATYPICAL CADHERIN 1 | FAT1-related glomerulo-tubular nephropathy | [4] |
| FAT4 | FAT ATYPICAL CADHERIN 4 | Van Maldergem syndrome 2 | [4] |
| PAX2 | PAIRED BOX GENE 2 | Papillo-renal syndrome/FSGS | [4] |
| LMX1B | LIM HOMEOBOX TRANSCRIPTION FACTOR 1, BETA | Nail–patella syndrome | [4] |
| KANK1 | KN MOTIF- AND ANKYRIN REPEAT DOMAIN-CONTAINING PROTEIN 1 | Cerebral palsy | [4] |
First insights into the importance of disease reclassification in SRNS came in recent years from case reports describing examples of phenocopies mimicking idiopathic SRNS that led to erroneous treatment. In 2013, Fervenza [20] reported the case of an 18-year-old patient presenting with nephrotic-range proteinuria who had been previously diagnosed as minimal change disease and then as “biopsy-missed” FSGS. Indeed, during the course of the disease, the patient received a therapeutic trial with steroids and cyclosporine. Only after treatment failure, further reconsideration of laboratory and biopsy findings allowed the suspicion of Dent disease that was then confirmed by genetic analysis, not performed earlier, showing a frame-shifting mutation in the CLCN5 gene. A similar case was reported by Saida et al. [21].
Besides single case reports, the first systematic studies showing the role of mutations in other than common SRNS-causing genes, but clinically mimicking the phenotype, were performed in patients with mutations in the Alport syndrome-related gene COL4A [34]. Starting from the observation that patients with autosomal dominant Alport syndrome due to heterozygous mutations in COL4A3 and COL4A4 could present with a variable clinical phenotype, including isolated severe proteinuria with FSGS at kidney biopsy, Malone et al. [25] speculated that some patients receiving a generic diagnosis of FSGS could instead be mutations carriers in COL4A3 and COL4A4 and so be misclassified. Using WES, they tested a cohort of 70 families with a diagnosis of familial FSGS. In seven out 70 families, genetic testing showed heterozygous mutations in COL4A3 or COL4A4 segregating with disease manifestations. Interestingly, in all seven families, there were individuals with nephrotic-range proteinuria and histologic features of FSGS by light microscopy. As the authors reported, these findings demonstrated a possible overlap between phenotypes induced by COL4A3 and COL4A4 variants and familial FSGS genes, suggesting that screening for rare variants/mutations in these genes in families referred with a diagnosis of familial FSGS is essential for better disease definition and treatment [25]. Similar results came from other studies where the genetic approach reclassified the diagnosis or the clinical suspicion of SRNS/FSGS as Alport syndrome spectrum [14][35][31][32]. Interestingly, some of these studies included also sporadic cases, demonstrating that the correct genetic diagnosis and disease reclassification are mandatory even in the absence of clear familial history [14]. In addition, attempts to identify subtle clinical findings related to the genetic diagnosis of Alport syndrome (e.g., glomerular basement membrane thinning/lamellation at electron microscopy) were sometimes performed, although not systematically [31][32]. A similar story applies to other genes, such as PAX2 [23].
In the last few years, thanks to the recognition that mutations in many genes can mimic SRNS, therefore acting as phenocopies, the spectrum of genes responsible for SRNS has been considerably widened. In a recent paper, Warejko et al. aimed at detecting monogenic causes of early-onset SRNS in an international cohort of 300 families by using WES and analysis of a large panel of genes including “phenocopy genes”. The authors reported detection of phenocopies in almost 5% of patients with SRNS. The mutations were found in eight phenocopy genes, specifically COL4A5, COL4A3, CLCN5, GLA, AGXT, CTNS, FN1 and WDR19 [3]. Interestingly, the term “phenocopy” was used for the first time in this work in the field of SRNS. Following a similar approach, phenocopies of monogenic podocytopathies can actually be found in other studies assessing the role of NGS in nephropathies, either in children or adults [7][15][18][33][30][36]. In these studies, unbiased extended genetic testing allowed authors to turn the initial clinical diagnosis into a specific genetic diagnosis. Specifically, phenocopies of monogenic podocytopathies were detected at a rate of 1–5%. All these patients received a generic clinical diagnosis of SRNS or FSGS before undergoing genetic analysis [7][14][15][33][30][36].
From a clinical point of view, the latest progress in the diagnostic algorithm is the possibility to couple extended genetic analysis with reverse phenotyping, enabling a more precise differentiation between overlapping phenotypes and leading to a reclassification of the diagnosis in individual patients [4][33][37]. Indeed, including genes responsible for inherited nephropathies other than monogenic podocytopathies (ideally, all “CKD genes”) represents the first step for a correct diagnosis of SRNS. However, the occurrence of unexpected findings needs to be addressed in order to provide reliable diagnostic results (Figure 1B). In a recent paper, Landini et al. systematically addressed this issue [4]. The diagnostic algorithm coupled WES for an extended panel of 298 genes related to CKD (including, but not limited to, SRNS-related genes) and reverse phenotyping, namely the reevaluation of patients and their families after genetic testing, looking for previously overlooked clinical features of the underlying genetic diagnosis. This approach was applied to a cohort of 111 patients affected by early-onset NS, including 64 patients with SRNS. Of note, genetic testing included parents of probands, allowing for assessing the segregation of variants. According to the literature, disease-causing variants in podocytopathy genes were detected in 30% of patients. However, reverse phenotyping permitted identifying previously unrecognized clinical signs of an unexpected underlying genetic nephropathy mimicking SRNS in 18 out of 64 patients (28%), confirming the genetic diagnosis. Phenocopy genes identified were COL4A3, COL4A4, COL4A5, LAMB2, GLA, FAT1, FAT4, PAX2, CLCN5, CTNS, LMX1B and KANK1 [4]. Very recently, clinical revaluation on the light of genetic findings was performed in selected cases of large cohorts of patients [33][30], confirming the utility of moving back from genotype to phenotype in order to reclassify diseases (Figure 1B). Interestingly, similar results have been provided also for other inherited kidney diseases, such as tubulopathies and nephronophthisis [10][11][38].
The high diagnostic capability to reclassify the original diagnosis based on clinical suspicion has also been recently confirmed in a real-life clinical setting by Jayasingh et al., who reported the analysis of the clinical impact of genomic testing in patients with suspected monogenic kidney diseases. In this work, NGS reclassified the clinical diagnosis with direct impact on subsequent clinical management and counseling [30].
References
- Report, North American Pediatric Renal Trials and Collaborative Studies: NAPRTCS Annual Transplant. Available online: http://we.emmes.com/study/ped/annlrept/annualrept2014.pdf (accessed on 1 September 2020).
- Becherucci, F.; Roperto, R.M.; Materassi, M.; Romagnani, P. Chronic kidney disease in children. Clin. Kidney J. 2016, 9, 583–591.
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62.
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse Phenotyping after Whole-Exome Sequencing in Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100.
- Szabó, T.; Orosz, P.; Balogh, E.; Jávorszky, E.; Máttyus, I.; Bereczki, C.; Maróti, Z.; Kalmár, T.; Szabo, A.J.; Reusz, G.; et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr. Nephrol. 2018, 33, 1713–1721.
- Van Der Ven, A.T.; Connaughton, D.M.; Ityel, H.; Mann, N.; Nakayama, M.; Chen, J.; Vivante, A.; Hwang, D.-Y.; Schulz, J.; Braun, D.A.; et al. Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2018, 29, 2348–2361.
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151.
- Daga, A.; Majmundar, A.J.; Braun, D.A.; Gee, H.Y.; Lawson, J.A.; Shril, S.; Jobst-Schwan, T.; Vivante, A.; Schapiro, D.; Tan, W.; et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018, 93, 204–213.
- Gee, H.Y.; Otto, E.A.; Hurd, T.W.; Ashraf, S.; Chaki, M.; Cluckey, A.; Vega-Warner, V.; Saisawat, P.; Diaz, K.A.; Fang, H.; et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014, 85, 880–887.
- Snoek, R.; Van Setten, J.; Keating, B.J.; Israni, A.; Jacobson, P.A.; Oetting, W.S.; Matas, A.J.; Mannon, R.B.; Zhang, Z.; Zhang, W.; et al. NPHP1 (Nephrocystin-1) gene deletions cause adult-onset ESRD. J. Am. Soc. Nephrol. 2018, 29, 1772–1779.
- Braun, D.A.; Schueler, M.; Halbritter, J.; Gee, H.Y.; Porath, J.D.; Lawson, J.A.; Airik, R.; Shril, S.; Allen, S.J.; Stein, D.; et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016, 89, 468–475.
- Armstrong, M.E.; Thomas, C.P. Diagnosis of monogenic chronic kidney diseases. Curr. Opin. Nephrol. Hypertens. 2019, 28, 183–194.
- Mann, N.; Braun, D.A.; Amann, K.; Tan, W.; Shril, S.; Connaughton, D.M.; Nakayama, M.; Schneider, R.; Kitzler, T.M.; Van Der Ven, A.T.; et al. Whole-exome sequencing enables a precision medicine approach for kidney transplant recipients. J. Am. Soc. Nephrol. 2019, 30, 201–215.
- Lata, S.; Marasa, M.; Li, Y.; Fasel, D.A.; Groopman, E.; Jobanputra, V.; Rasouly, H.; Mitrotti, A.; Westland, R.; Verbitsky, M.; et al. Whole-exome sequencing in adults with chronic kidney disease: A pilot study. Ann. Intern. Med. 2018, 168, 100–109.
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; Van Der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928.
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146.
- Kopp, J.B.; Anders, H.-J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68.
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970.
- Pierides, A.; Voskarides, K.; Athanasiou, Y.; Ioannou, K.; Damianou, L.; Arsali, M.; Zavros, M.; Pierides, M.; Vargemezis, V.; Patsias, C.; et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transpl. 2009, 24, 2721–2729.
- Fervenza, F.C. A patient with nephrotic-range proteinuria and focal global glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1979–1987.
- Saida, K.; Kamijo, Y.; Matsuoka, D.; Noda, S.; Hidaka, Y.; Mori, T.; Shimojo, H.; Ehara, T.; Miura, K.; Takita, J.; et al. A case of adult Dent disease in Japan with advanced chronic kidney disease. CEN Case Rep. 2014, 3, 132–138.
- Edwards, N.; Rice, S.J.; Raman, S.; Hynes, A.M.; Srivastava, S.; Moore, I.; Al-Hamed, M.; Xu, Y.; Santibanez-Koref, M.; Thwaites, D.T.; et al. A novel LMX1B mutation in a family with end-stage renal disease of ‘unknown cause’. Clin. Kidney J. 2015, 8, 13–19.
- Barua, M.; Stellacci, E.; Stella, L.; Weins, A.; Genovese, G.; Muto, V.; Caputo, V.; Toka, H.R.; Charoonratana, V.T.; Tartaglia, M.; et al. Mutations in PAX2 associate with adult-onset FSGS. J. Am. Soc. Nephrol. 2014, 25, 1942–1953.
- He, G.; Zhang, H.; Cao, S.; Xiao, H.; Yao, Y. Dent’s disease complicated by nephrotic syndrome: A case report. Intractable Rare Dis. Res. 2016, 5, 297–300.
- Malone, A.F.; Phelan, P.J.; Hall, G.; Cetincelik, U.; Homstad, A.; Alonso, A.S.; Jiang, R.; Lindsey, T.B.; Wu, G.; Sparks, M.A.; et al. Rare hereditary COL4A3/COL4A4 variants maybe mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014, 86, 1253–1259.
- Vallés, P.; Peralta, M.; Carrizo, L.; Martin, L.; Principi, I.; Gonzalez, A.; Manucha, W. Follow-up of steroid-resistant nephrotic syndrome: Tubular proteinuria and enzymuria. Pediatr. Nephrol. 2000, 15, 252–258.
- Wang, X.; Anglani, F.; Beara-Lasic, L.; Mehta, A.J.; Vaughan, L.E.; Hernandez, L.H.; Cogal, A.; Scheinman, S.J.; Ariceta, G.; Isom, R.; et al. Glomerular pathology in Dent disease and its association with kidney function. Clin. J. Am. Soc. Nephrol. 2016, 11, 2168–2176.
- Du Moulin, M.; Koehn, A.; Golsari, A.; Dulz, S.; Atiskova, Y.; Patten, M.; Münch, J.; Avanesov, M.; Ullrich, K.; Muschol, N. The mutation p.D313Y is associated with organ manifestation in Fabry disease. Clin. Genet. 2017, 92, 528–533.
- Köping, M.; Shehata-Dieler, W.; Cebulla, M.; Rak, K.; Oder, D.; Müntze, J.; Nordbeck, P.; Wanner, C.; Hagen, R.; Schraven, S. Cardiac and renal dysfunction is associated with progressive hearing loss in patients with Fabry disease. PLoS ONE 2017, 12, e0188103.
- Jayasinghe, K.; Dm, Z.S.; Kerr, P.G.; Gaff, C.; Martyn, M.; Whitlam, J.; Creighton, B.; Bn, E.D.; Hunter, M.; Jarmolowicz, A.; et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet. Med. 2020.
- Xie, J.; Wu, X.; Ren, H.; Wang, W.; Pan, X.; Hao, X.; Tong, J.; Ma, J.; Ye, Z.; Meng, G.; et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell Biol. 2014, 6, 498–505, Erratum in 2015, 7, 184.
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223.
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. Am. J. Kidney Dis. 2020, 76, 460–470.
- Torra, R.; Furlano, F.; Ars, E. How genomics reclassifies diseases: The case of Alport syndrome. Clin. Kidney J. 2020, 1–3.
- Adam, J.; Connor, T.M.; Wood, K.; Lewis, D.; Naik, R.; Gale, D.P.; Sayer, J.A. Genetic testing can resolve diagnostic confusion in Alport syndrome. Clin. Kidney J. 2014, 7, 197–200.
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, Á.; Ariceta, G.; Borregán, M.; et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371.
- De Haan, A.; Eijgelsheim, M.; Vogt, L.; Knoers, N.V.A.M.; de Borst, M.H. Diagnostic yield of next-generation sequencing in patients with chronic kidney disease of unknown etiology. Front. Genet. 2019, 10, 1264.
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Özen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101.


