 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Le Minh Tu Phan | + 1433 word(s) | 1433 | 2020-11-17 04:53:49 | | | |
| 2 | Nicole Yin | Meta information modification | 1433 | 2020-11-17 11:16:08 | | |
Video Upload Options
Core biomarkers amyloid beta (Aβ) and Tau have been considered as key neuropathological hallmarks of Alzheimer's disease. However, they did not sufficiently reflect clinical severity and therapeutic response, proving the difficulty of the Aβ- and Tau-targeting therapies in clinical trials. Along with these core biomarkers, non-Amyloidbeta-Tau pathophysiological biomarkers (Neurodegeneration-related biomarkers, biomarkers for neuroinflammation and phagocytosis of an innate immune system, lipid metabolism biomarkers) could serve as advanced reporters for early diagnosing AD, predicting AD progression, and monitoring the treatment response.
1. Introduction
Alzheimer’s disease (AD) is characterized as a progressive neurodegenerative disorder that causes memory deficits and cognitive impairment. Pathologically, AD is associated with the formation of senile plaques and neurofibrillary tangles in the brain by the accumulations of aggregated amyloid-β (Aβ) and Tau proteins, which are considered as central hallmarks in AD[1][2][3].
2. Importance of Non-Aβ-Tau Biomarkers in Monitoring Alzheimer’s Disease
Currently, AD diagnoses are having to face enormous challenges in which the clinical symptoms occur decades after accumulating neuropathological modifications[4]. It is well known that extracellular Aβ deposition and the intracellular hyperphosphorylation of Tau proteins are general considerations for AD’s diagnostic biomarkers and various hypotheses have been put forth to shed light on the pathogenesis from multi-omics studies[4][5]. Aβ monomers generally consist of 36–43 amino acids; however, the Aβ42/40 ratios in CSF, usually measured by immunoassays or Aβ positron emission tomography (PET) imaging are most broadly evaluated that reflect Aβ aggregation and subsequent senile plaques formation[5][6][7]. In parallel with amyloidosis, Tau, a microtubule-binding protein phosphorylated and accumulated into neurofibrillary tangles (NFT), is reflected as a second biomarker for AD[7]. In terms of AD prediction, total Tau (T-tau), as well as Tau phosphorylated at threonine 181 (P-tau), are the core CSF predictors[8]. In normal physiological conditions, Aβ functions to regulate learning and memory, neurogenesis, angiogenesis and repair leaks in the blood–brain barrier (BBB), etc., while Tau protein also holds several nerve-related essential roles such as myelination, axonal transport, neuronal excitability, microtubule dynamics, so on[9]. Nevertheless, various in vitro studies revealed that upon the challenge of synthetic Aβ42, the observable results in the human induced pluripotent stem cell iPSC-derived neuron demonstrated several neuronal deficits such as neuronal death, ER stress or synaptotoxicity[10]. Furthermore, the high Aβ42/40 ratio can robustly induce Tau hyperphosphorylation and perhaps neurodegeneration[10]. In turn, Mclnnes’s group indicated that the interaction between Tau and synaptogyrin-3 lessened synaptic neurotransmitter release, as well as attenuated protein translation and nuclear transcription, consequently associated with neuronal dysfunction and cognitive decline[11]. From these reasons, Aβ and Tau species are the main targets of numerous studies to develop biosensors that allow the detection in both invasive samples such as CSF, plasma[12][13][14][15] and non-invasive samples such as saliva and urine[16][17].
Based on conventional understanding about AD pathology, numerous laboratory studies and clinical trials made intensive attempts to disrupt the refractory of AD via Aβ and Tau targeting[6]. Many studies are under different phases of evaluation; unfortunately, almost completed ones have been comprehensively futile because of facing primary cognitive outcomes, especially in phase III trial[6][18]. To further investigate an efficacious therapeutic target, remaining pathological alterations in the brain were considered, including inflammation, neurodegeneration, lipid metabolism, synaptic dysfunction, protein clearance, and mitochondrial dysfunction[7][19][20][21][22][23]. These modifications directly regulate preclinical AD toward persistent and multifaceted AD dementia[4]. Therefore, molecules associated with the multifaceted nature of AD pathophysiological progression have been considered as novel biomarkers in AD (Figure 1).
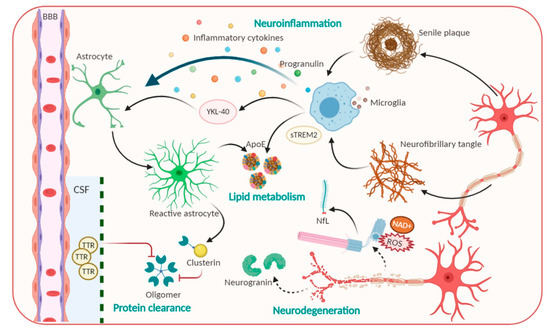
Figure 1. Pathophysiological processes including Amyloid beta, Tau and candidate non-Aβ-Tau biomarkers for Alzheimer’s disease.
Neurodegeneration-related biomarkers: New promising candidates for AD diagnosis. Neurodegeneration is not only inevitable but also exacerbated in AD progression[23], as various neuronal and synaptic-related proteins which are most associated with brain development have been suggested to be involved in the first step of AD progression, and their function precedes neuronal loss, thus allowing them to be considered as CSF biomarkers for AD. Typically, visinin-like protein 1 (VLP-1) can seep out from dented neurons and act as a vital calcium sensor protein. VLP-1 was shown to be significantly increased in AD, suggesting it as a useful biomarker that correlates with the degree of dementia. Currently, combined analyses of Aβ, P-tau, and VLP-1 have been performed and were reported to increase the accuracy of AD diagnosis[24][25]. Furthermore, growth-associated protein, which is another synaptic protein involved in the regulation of axonal outgrowth, synaptic plasticity, and learning and memory functions, was found to be present at higher levels in CSF[24][26]. Particularly, neurofilament light (NfL) polypeptide, an axonal cytoskeleton composition, is leaked from axonal injury into brain interstitial fluid, then tracked into CSF and blood[6][27]. Previous studies reported that NfL concentration is elevated approximately 16 years before the judgment of disease onset. Measuring the NfL level can be taken place in CSF and blood samples for hypometabolism and neurodegeneration, especially with changing cognitive scores. For these reasons, NfL elevated rates express as a great feature for the cost-effective and non-invasive diagnostic measurement of a broad range of neurodegeneration diseases, as well as clinical progression in pre-symptomatic of AD[6][28].
Neuroinflammation and phagocytosis of an innate immune system: Potential therapeutic targets. The propagation of phagocytosis and the inflammatory process, which are involved in the initiation and exacerbation of AD, are among the most attractive events for AD physiological behavior identification[29][30]. Indeed, microglia—brain resident macrophages—are responsible for microenvironmental surveillance, the clearance of debris and pathogens, and sustaining the secretion of proinflammatory mediators[29]. Additionally, conclusive evidence demonstrated that inflamed molecules, such as those in iNOS production, tend to speed up Αβ aggregation and senile plaques formation, ultimately leading to a precarious vicious cycle[29][31]. Of note, a triggering receptor expressed on myeloid cells 2 (TREM2), which is highly expressed in microglia, modulates plaque-surrounding microglial activities including survival, proliferation, cytokine release as well as biosynthetic metabolism[32]. Nevertheless, compelling evidence has revealed that the levels of the ectodomain of TREM2, which was proteolytic cleaved and liberated to generate extracellular soluble TREM2 (sTREM2), were elevated in the CSF in AD stage-dependent milieu[32][33]. sTREM2 not only recapitulated full-length TREM2-like functions but also contributes to recruiting microglia to the plaques. Significantly, Ewers group’s outcomes denoted that higher CSF sTREM2 levels are responsible for less cognitive decline in hippocampal volume[32][33]. Accordingly, higher CSF sTREM2 concentration may act as a biomarker representing the amelioration of pathological progression at the AD’s symptomatic stage[33]. Besides microglia, another star-shaped glial cell—astrocytes—also play essential roles in Aβ phagocytosis and degradation, strengthening trophic nerves as well as generating a safety barrier between Aβ accumulation and neurons. However, a result reported that upon the chronic stress, astrocytes overexpress β-secretase (BACE1), which induce Aβ overproduction[34]. β2-microglobulin, intercellular adhesion molecule 1 (ICAM1), progranulin and chitinase-3-like protein 1 (CHI3L1/YKL-40) also participate in neuroinflammation, thus, affecting AD pathology[23]. YKL-40 was expressed in activated astrocytes and microglia[20] whose level is associated with an enhanced early AD continuum and exacerbated neuroinflammation; thus, it exerts the features of a promising biomarker for AD[4]. Furthermore, molecules related to the uptake and degradation of unfolded Aβ and hyperphosphorylated Tau, have received much more interest as potential biomarkers. Typically, transthyretin (TTR) or clusterin, are those that are elevated in CSF, and act as a molecular chaperon that can directly bind to the Aβ molecule to prevent Aβ accumulation and the resultant attenuated Aβ-associated cellular toxicity[4][23]. Hence, these factors perform protective activities against the excessive Aβ load, thereby serving as a potential candidate for stage and state AD diagnosis.
Lipid metabolism biomarkers. Lipid metabolites are highly associated with AD progression; thus, they have been investigated as promising disease biomarkers[22]. The first biomarker that markedly increases the risk for developing AD is ApoE, the molecule that is involved in the normal catabolism of triglyceride-rich lipoproteins and exhibits immunoreactivity in Aβ deposits and NFTs. ApoE is a glycoprotein that is highly expressed in the brain. This glycoprotein contains 299 amino acids and is classified into three common isoforms in humans that differ in their structures[35][36]. ApoE regulates the isoform-dependent removal of Aβ, via Aβ lipoprotein complexes endocytosis, by influencing proteolytic degradation of Aβ and facilitating its transport across BBB[4]. In addition, ApoE has been shown to influence microglial activation states and cellular responses in a TREM2-dependent way; especially ApoE-knockdown in mice blocks microglial phagocytic function to Aβ[37]. Numerous studies imply that ApoE4 harmfully accelerates Aβ aggregation by interacting with Aβ to promote Aβ aggregation and to stabilize Aβ oligomers. On the other hand, other pieces of evidence showed that ApoE2 exerts a protective function in AD[38]. Therefore, the quantification of isoform-dependent ApoE levels is promising as a CSF diagnostic biomarker.
References
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216.
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell. Mol. Life Sci. 2019, 76, 1833–1863.
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019, 51, 1–10.
- Zetterberg, H.; Schott, J.M. Biomarkers for Alzheimer’s disease beyond amyloid and tau. Nat. Med. 2019, 25, 201–203.
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217.
- Ashton, N.J.; Ide, M.; Zetterberg, H.; Blennow, K. Salivary Biomarkers for Alzheimer’s Disease and Related Disorders. Neurol. Ther. 2019, 8, 83–94.
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447.
- Sang Su Kwak; Kevin J. Washicosky; Emma Brand; Djuna Von Maydell; Jenna Aronson; Susan Kim; Diane E. Capen; Murat Cetinbas; Ruslan Sadreyev; Shen Ning; et al.Enjana BylykbashiWeiming XiaSteven L. WagnerSe Hoon ChoiRudolph E. TanziDoo Yeon Kim Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer's disease.. Nature Communications 2020, 11, 1377, 10.1038/s41467-020-15120-3.
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193.
- Devi, R.; Gogoi, S.; Dutta, H.S.; Bordoloi, M.; Sanghi, S.K.; Khan, R.J.N.A. Au/NiFe2O4 nanoparticle-decorated graphene oxide nanosheets for electrochemical immunosensing of amyloid beta peptide. Nanoscale Adv. 2020, 2, 239–248.
- Karaboga, M.N.S.; Sezgintürk, M.K. Analysis of Tau-441 protein in clinical samples using rGO/AuNP nanocomposite-supported disposable impedimetric neuro-biosensing platform: Towards Alzheimer’s disease detection. Talanta 2020, 219, 121257.
- Kim, K.; Park, C.B.J.B. Femtomolar sensing of Alzheimer’s tau proteins by water oxidation-coupled photoelectrochemical platform. Biosens. Bioelectron. 2020, 154, 112075.
- Liu, B.; Shen, H.; Hao, Y.; Zhu, X.; Li, S.; Huang, Y.; Qu, P.; Xu, M.J.A.c. Lanthanide Functionalized Metal–Organic Coordination Polymer: Toward Novel Turn-On Fluorescent Sensing of Amyloid β-Peptide. Anal. Chem. 2018, 90, 12449–12455.
- Altuntas, S.; Buyukserin, F. Fabrication of thioflavin-T-modified nanopillared SERS substrates for ultrasensitive beta-amyloid peptide detection. J. Raman Spectrosc. 2018, 49, 1247–1256.
- Chan, H.-N.; Xu, D.; Ho, S.-L.; Wong, M.S.; Li, H.-W.J.C.S. Ultra-sensitive detection of protein biomarkers for diagnosis of Alzheimer’s disease. Chem. Sci. 2017, 8, 4012–4018.
- Jeffrey Cummings; Garam Lee; Aaron Ritter; Kate Zhong; Alzheimer's disease drug development pipeline: 2018. Alzheimer's & Dementia: Translational Research & Clinical Interventions 2018, 4, 195-214, 10.1016/j.trci.2018.03.009.
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; Dobricic, V.; et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimer’s Dement. 2019, 15, 776–787.
- Cummings, J. The role of biomarkers in Alzheimer’s disease drug development. In Reviews on Biomarker Studies in Psychiatric and Neurodegenerative Disorders; Springer: Berlin/Heidelberg, Germany, 2019; pp. 29–61.
- Zetterberg, H. Blood-based biomarkers for Alzheimer’s disease—An update. J. Neurosci. Methods 2019, 319, 2–6.
- Peña-Bautista, C.; Baquero, M.; Vento, M.; Cháfer-Pericás, C. Free radicals in Alzheimer’s disease: Lipid peroxidation biomarkers. Clin. Chim. Acta 2019, 491, 85–90.
- Park, S.A.; Han, S.M.; Kim, C.E. New fluid biomarkers tracking non-amyloid-β and non-tau pathology in Alzheimer’s disease. Exp. Mol. Med. 2020, 52, 556–568.
- Anoop, A.; Singh, P.K.; Jacob, R.S.; Maji, S.K. CSF Biomarkers for Alzheimer’s Disease Diagnosis. Int. J. Alzheimer’s Dis. 2010, 2010, 606802.
- Lee, J.-M.; Blennow, K.; Andreasen, N.; Laterza, O.; Modur, V.; Olander, J.; Gao, F.; Ohlendorf, M.; Ladenson, J.H. The Brain Injury Biomarker VLP-1 Is Increased in the Cerebrospinal Fluid of Alzheimer Disease Patients. Clin. Chem. 2008, 54, 1617–1623.
- Åsa Sandelius; Erik Portelius; Åsa Källén; Henrik Zetterberg; Uros Rot; Bob Olsson; Jon B. Toledo; Leslie M. Shaw; Virginia M. Y. Lee; David J. Irwin; et al.Murray GrossmanDaniel WeintraubAlice Chen-PlotkinDavid A. WolkLeo McCluskeyLauren ElmanVesna KostanjeveckiManu VandijckJennifer McBrideJohn Q. TrojanowskiKaj Blennow Elevated CSF GAP-43 is Alzheimer's disease specific and associated with tau and amyloid pathology. Alzheimer's & Dementia 2018, 15, 55-64, 10.1016/j.jalz.2018.08.006.
- Henrik Zetterberg; Samantha C. Burnham; Blood-based molecular biomarkers for Alzheimer’s disease. Molecular Brain 2019, 12, 1-7, 10.1186/s13041-019-0448-1.
- Oliver Preische; Dominantly Inherited Alzheimer Network; Stephanie A. Schultz; Anja Apel; Jens Kuhle; Stephan A. Kaeser; Christian Barro; Susanne Gräber; Elke Kuder-Buletta; Christian Lafougere; et al.Christoph LaskeJonathan VögleinJohannes LevinColin L. MastersRalph MartinsPeter R. SchofieldMartin N. RossorNeill R. Graff-RadfordStephen SallowayBernardino GhettiJohn M. RingmanJames M. NobleJasmeer ChhatwalAlison M. GoateTammie L. S. BenzingerJohn C. MorrisRandall J. BatemanGuoqiao WangAnne M. FaganEric M. McDadeBrian A. GordonMathias Jucker Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nature Medicine 2019, 25, 277-283, 10.1038/s41591-018-0304-3.
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; García-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennström, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 251–273.
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532.
- Jack Van Horssen; Pauline Van Schaik; Maarten Witte; Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders?. Neuroscience Letters 2019, 710, 132931, 10.1016/j.neulet.2017.06.050.
- Li Zhong; Ying Xu; Rengong Zhuo; Tingting Wang; Kai Wang; Ruizhi Huang; Daxin Wang; Yue Gao; Yifei Zhu; Xuan Sheng; et al.Kai ChenNa WangLin ZhuDan CanYuka MartenMitsuru ShinoharaChia-Chen LiuDan DuHao SunLei WenHuaxi XuGuojun BuXiao-Fen Chen Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nature Communications 2019, 10, 1-16, 10.1038/s41467-019-09118-9.
- Michael Ewers; Nicolai Franzmeier; Marc Suárez-Calvet; Estrella Morenas-Rodriguez; Miguel Angel Araque Caballero; Gernot Kleinberger; Laura Piccio; Carlos Cruchaga; Yuetiva Deming; Martin Dichgans; et al.John Q. TrojanowskiLeslie M. ShawMichael W. WeinerChristian HaassAlzheimer’S Disease Neuroimaging Initiative Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Science Translational Medicine 2019, 11, eaav6221, 10.1126/scitranslmed.aav6221.
- Jose Miguel Rubio-Perez; Juana Maria Morillas-Ruiz; A Review: Inflammatory Process in Alzheimer's Disease, Role of Cytokines. The Scientific World Journal 2012, 2012, 1-15, 10.1100/2012/756357.
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056.
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE Isoforms Differentially Regulate Brain Amyloid- Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57.
- Aivi T. Nguyen; Kui Wang; Gang Hu; Xuran Wang; Zhen Miao; Joshua A. Azevedo; EunRan Suh; Vivianna M. Van Deerlin; David Choi; Kathryn Roeder; et al.Mingyao LiEdward B. Lee APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathologica 2020, 140, 1-17, 10.1007/s00401-020-02200-3.
- Takahisa Kanekiyo; Huaxi Xu; Guojun Bu; ApoE and Aβ in Alzheimer’s Disease: Accidental Encounters or Partners?. Neuron 2014, 81, 740-754, 10.1016/j.neuron.2014.01.045.


