 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Barbaros Eroglu | + 2740 word(s) | 2740 | 2019-12-04 02:10:35 | | | |
| 2 | Barbaros Eroglu | + 36 word(s) | 2776 | 2020-11-06 08:27:07 | | | | |
| 3 | Veysel Kayser | Meta information modification | 2776 | 2021-09-21 04:58:52 | | |
Video Upload Options
We provide an overview of the challenges and advancements in the development of therapeutic monoclonal antibodies and antibody products.
Development of Therapeutic Monoclonal Antibodies
Barbaros Eroglu1, Veysel Kayser1*, Vicki Sifniotis1, Esteban Cruz1
1 Sydney School of Pharmacy, Faculty of Medicine and Health, The University of Sydney
*Correspondence: veysel.kayser@sydney.edu.au
1. Introduction
Since the first therapeutic monoclonal antibody (mAb) was approved by the USA Food and Drug Administration in 1986, full-length monoclonal antibodies have become and persistently remain the most dominant and significant class of biologics in the pharmaceutical industry due to their unmatched specificity and their ability to treat a variety of diseases including cancers, infections, autoimmune disorders and cardiovascular and neurological diseases (1-3).
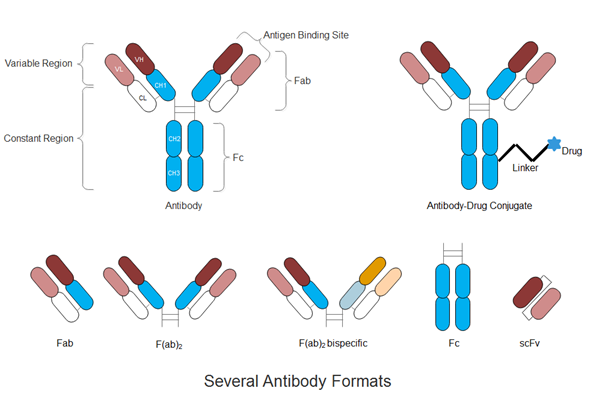
In addition to the dominant full-length monoclonal antibodies, antibody (Ab) engineering technologies have progressed in recent years to produce highly optimized, strategically engineered biobetter therapies (4-6). Fragments of mAbs such as the crystallizable fragment (Fc), antigen binding fragment (Fab), and single-chain variable fragment (scFv) possess key functions including specificity to a biological target or immunological activation. The isolation of these fragments for fusion with other mAb fragments, biologically functional proteins, cytotoxic drugs, or drug carriers has been the main interest in developing the next generation of biobetter therapies such as antibody-drug-conjugates, nanoparticle-antibody complexes and bispecifics (5, 7-10).
Figure 1. Schematic illustration of full-length antibody, antibody-drug conjugate and several antibody formats
2. Key Challenges and Considerations in Antibody Development
a) Challenges in Discovery
Full-length antibodies are large molecules comprising of more than 1000 amino acids forming four peptide chains (two heavy and two light-chains) that are bound together by interchain disulfide bonds and interchain non-covalent interactions. Due to their sizes, chemical synthesis is not feasible for production of such large proteins.
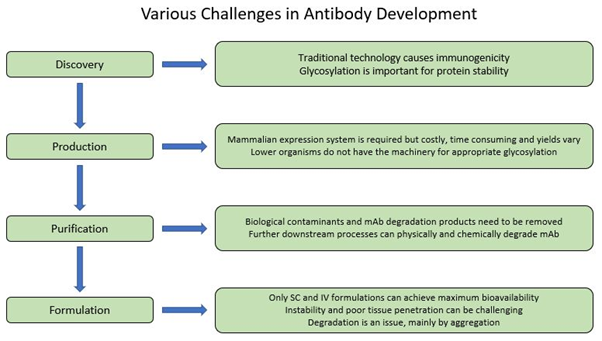
First-generation technologies for full-length mAb discovery required the immunization of animals, which resulted in the generation of highly specific antibody libraries of non-human origin that were potentially immunogenic and less effective in inducing an immune response as compared to wholly human mAbs (5, 7, 8). In addition, several studies have shown that some plant species such as tobacco can be used to express full-length mAbs; however, immunogenicity and lack of human glycosylation is still an issue that needs to be addressed in these systems (11-13).
b) Challenges in Production and Purification
A mammalian expression system is required to produce a full-length antibody since lower organisms do not have the necessary machinery for the production. Bacteria, for example do not have the tools for post-translational glycosylation, and only few bacteria species have the capacity form disulfide bonds, which are essential components of antibodies. Yeasts, on the other hand, can glycosylate but with high amount of mannose which can cause immunogenicity (5, 12).
Utilizing mammalian expression systems also has some pitfalls as it is considerably costly and inefficient, can have varied yields depending on the product and expression system, and requires additional downstream processing to purify and remove biological contaminants introduced from the expression system (5, 14, 15).
c) Formulation Challenges
Another challenge with mAbs and Ab-based therapeutics is the limitation to lyophilised and liquid-based formulations for intravenous (IV) or subcutaneous (SC) delivery at high protein concentrations to achieve maximum bioavailability. Pulmonary delivery creates additional mechanical stress that contributes to instability of the mAb but more critically, systemic delivery of large proteins through respiratory system is quite limited, and oral delivery is unfeasible because of chemical and enzymatic degradation and limited absorption in the gastric and intestinal system (5, 16, 17). Therefore, mAbs and other Ab therapeutics are formulated as injectables and constituted using various stabilizing additives such as sugars, salts, surfactants and buffered to achieve a certain solution pH range.
d) Other Challenges
Aggregation and instability are two other critical obstacles in antibody development. The aggregation profile of antibodies can be influenced by both external conditions such as temperature and purification procedure and intrinsic factors. Stability of antibodies can also be affected by other parameters such as amino acid composition and protein concentration. All these issues need to be addressed as they have a great impact on pharmaceutical properties including formulation, route of administration and biological half-life (18-21). Prediction of antibody degradation and in turn shelf-life calculation based on accelerated studies remains a challenge, nevertheless new methods are discussed to overcome this problem (22, 23).
Figure 2. Schematic diagram of various challenges in antibody development process.
3. Advancements in Antibody Discovery and Production Processes
Utilizing immunized animals for full-length mAb discovery resulted in the generation of fully murine mAbs which can be immunogenic. Technologies arose to address immunogenicity, such as producing chimeric and humanized antibodies and developing transgenic mice with their murine antibody heavy and light chain genes replaced with equivalent human genes (5, 7, 8). Phage display technology; a method which involves the expression of proteins on the surface of phages to select desirable antibody variable domains based on their binding properties, is one of the commonest methods to produce fully human antibodies. The first approved human antibody, adalimumab, was discovered using phage display technology and it is the top selling pharmaceutical in the market with sales of nearly $20 billion in 2018 (1, 5, 24).
Significant variations in expression, yield, and quality of a mAb in hybridoma cell lines led to the development of high yielding mammalian expression systems, which utilize stable Chinese hamster ovary (CHO), mouse myeloma (NS0), and mouse hybridoma (Sp2/0) cell lines (5, 7). In the context of research and development, transient expression in human cell lines including embryonic kidney (HEK 293), amniotic (CAP), a hybrid of HEK 293 and lymphoma (HKB-11), and embryonic retina (PER.C6) are favoured over stable CHO expression due to the ease and speed of production of workable quantities of antibody for preliminary studies. Although these expression systems might become dominant in the future, CHO cell lines are still the most widely used expression system in the industry as they can easily grow in suspension culture and ensure reproducibility between different batches (5, 7, 12, 25-27).
Another limitation of utilizing CHO expression systems is that, because of their different glycosylation profile, CHO-expressed mAbs may produce immunogenic non-human glycoform (Neu5Gc), and may have a higher composition of sialylation, which may result in reduced antibody-dependent cellular cytotoxicity (12, 25, 26). Approval of mAb Fc fusion therapeutics which are manufactured in HEK 293 expression system promotes the acceptance of human-based expression systems for the production of mAb-based therapeutics (7).
Although considered high risk due to potential endotoxin contamination in the product, lower organism expression platforms such as E. coli and P. pastoris continue to be the preferred option for the manufacture of fragment mAb formats owing to the relative ease, high yield, and reduced cost for manufacture as compared to mammalian expression platforms. The Fragment mAb formats produced in these systems are small in size and do not require glycosylation (5, 12, 28).
An emerging in vitro cell-free synthesis technology is being developed with bacterial and CHO cell lysates which has the potential to alleviate formation of undesirable biological byproducts, since the machinery from the cell lysates purely express protein from the mAb genes that are introduced to the system. Despite not being currently applicable for industrial scale manufacture, this technology holds much promise as an alternative to live culture (29-32).
Protein A-based affinity chromatography is the most robust, efficient, and prevalently used mAb purification technology not only because of its selectivity and high affinity towards Fc regions of various species, but also its efficient dissociation of captured mAbs at a low pH for reuse. Once mAb is captured, further polishing steps may be required to remove further contaminants including host cell proteins and DNA, as well as leached affinity chromatography ligand- and Ab-degradation products such as fragments, aggregates, and ionic variants. Ion Exchange Chromatography (IEC), Size Exclusion Chromatography (SEC) and Hydrophobic Interaction Chromatography (HIC) are the most widely used polishing methods (33, 34).
4. Formulation Strategies and Improving Tissue Penetration
Strategies to improve the formulation of mAbs and Ab-based therapies continues to be an ongoing challenge, as is faced with all biotherapeutics. Despite the majority of currently approved mAb therapeutics being formulated for IV delivery, the interest in developing injectable formulations, primarily SC for systemic delivery, has increased owing to their advantages over IV delivery such as reduced burden to allied health services, patient tolerance, and adherence to treatment (35, 36). Nevertheless, certain mAb therapies are suitable, and others are not based on their solubility, viscosity, self-association, intrinsic stability, aggregation, and precipitation profiles. To address these issues; use of excipients, for instance, polysorbates, human hyaluronidase, amino acids, ionic liquids can be considered in the formulation; however, high concentrations of polysorbates can cause denaturation of proteins, and addition of human hyaluronidase can add burden to the viscosity of the formulation (5, 20, 37-41).
Delivery of the mAbs and Ab-products to the specific target tissue, for instance, tumors for cancer treatments, continues to be a challenge as penetration to tissue from blood vessels is poor, owing to mAbs’ polar surface charge and relatively large MW, limiting transport through physiological barriers(4, 6, 42). The enhancement of the affinity of mAbs to their biological target through strategic mutation technologies improved diffusion rates of mAb to target for increased efficacy. In the case of tumors, however, very high affinity can decrease diffusion due to the ``binding site barrier``. Increased affinity of mAbs results in enhanced internalization of mAbs and this mechanism is exploited through the development of high affinity antibody-drug conjugates (ADC) to target the delivery and release of potent drugs, inducing targeted cell death (42-45).
Fragment mAb platforms have shown a better tissue penetration and biodistribution than full-length antibodies; however, smaller peptides lacking an Fc region demonstrated highly reduced in vivo half-life and poor retention times. PEGylation is the primary strategy to address this issue, along with strategic Fc mutation and glycosylation engineering (21). Contrarily, relatively larger nanocarrier platforms have demonstrated superior pharmacokinetics and tumour retention, as well as enhanced stability and the sustained, controlled release of mAbs. Further to nanocarriers, additional formulation strategies developed for the controlled release of mAbs include hydrogels and crystalline antibodies, which have shown success as stable, injectable formulations for development (5, 10, 20, 43, 45, 46).
5. Future Outlook
Antibodies have been the most successful biologic therapeutic for several years and with the advancements in the technology and increased research on developing biosimilars and new antibody formats, it is clear that antibodies and their formats will remain the most thriving class of biopharmaceuticals in the forthcoming years. By making use of the new advancements in antibody development including cell-free synthesis methods, fully humanized antibody development methods, utilizing novel excipients for better formulations and different routes of administration, it will be possible to produce different types of antibodies and formats with relative ease, higher yield and less immunogenicity concern. With the help of the discovery and increased production of different antibody formats such as bispecifics, nanoparticle-antibody complexes and ADCs, the possibility of targeting and treating a variety of diseases will be much higher than ever before.
6. Acknowledgement
We thank Dr Mouhamad Reslan for critical reading of the manuscript and providing comprehensive feedback.
REFERENCES
- Urquhart L (2019) Top drugs and companies by sales in 2018. Nat Rev Drug Discov.
- Ecker DM, Jones SD, & Levine HL (2015) The therapeutic monoclonal antibody market. mAbs 7(1):9-14.
- An Z (2018) "Magic Bullets" at the center stage of immune therapy: a special issue on therapeutic antibodies. Protein Cell 9(1):1-2.
- Sifniotis V, Cruz E, Eroglu, & Kayser V (2019) Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies 8:36.
- Elgundi Z, Reslan M, Cruz E, Sifniotis V, & Kayser V (2017) The state-of-play and future of antibody therapeutics. Advanced Drug Delivery Reviews 122:2-19.
- Cruz E & Kayser V (2019) Monoclonal antibody therapy of solid tumors: clinical limitations and novel strategies to enhance treatment efficacy. Biologics 13:33-51.
- Santos MLd, Quintilio W, Manieri TM, Tsuruta LR, & Moro AM (2018) Advances and challenges in therapeutic monoclonal antibodies drug development. Brazilian Journal of Pharmaceutical Sciences 54.
- Almagro JC, Daniels-Wells TR, Perez-Tapia SM, & Penichet ML (2018) Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Frontiers in Immunology 8(1751).
- Strohl WR (2018) Current progress in innovative engineered antibodies. Protein & Cell 9(1):86-120.
- Yu M, Wu J, Shi J, & Farokhzad OC (2016) Nanotechnology for protein delivery: Overview and perspectives. Journal of Controlled Release 240:24-37.
- Pujol M, et al. (2007) Fighting cancer with plant-expressed pharmaceuticals. Trends in Biotechnology 25(10):455-459.
- Mizukami A, Caron AL, Picanço-Castro V, & Swiech K (2018) Platforms for Recombinant Therapeutic Glycoprotein Production. Recombinant Glycoprotein Production: Methods and Protocols, eds Picanço-Castro V & Swiech K (Springer New York, New York, NY), pp 1-14.
- Komarova TV, et al. (2011) Plant-made trastuzumab (herceptin) inhibits HER2/Neu+ cell proliferation and retards tumor growth. PLoS One 6(3):e17541.
- Müller-Späth T & Morbidelli M (2014) Purification of Human Monoclonal Antibodies and Their Fragments. Human Monoclonal Antibodies: Methods and Protocols, ed Steinitz M (Humana Press, Totowa, NJ), pp 331-351.
- Arosio P, Barolo G, Müller-Späth T, Wu H, & Morbidelli M (2011) Aggregation Stability of a Monoclonal Antibody During Downstream Processing. Pharmaceutical Research 28(8):1884-1894.
- Morales JO, et al. (2017) Challenges and Future Prospects for the Delivery of Biologics: Oral Mucosal, Pulmonary, and Transdermal Routes. The AAPS journal 19(3):652-668.
- Bittner B, Richter W, & Schmidt J (2018) Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs 32(5):425-440.
- Lu Z-J, et al. (2012) Frontier of therapeutic antibody discovery: The challenges and how to face them. World journal of biological chemistry 3(12):187-196.
- Kayser V, et al. (2011) Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnology Journal 6(1):38-44.
- Awwad S & Angkawinitwong U (2018) Overview of Antibody Drug Delivery. Pharmaceutics 10(3):83.
- Reslan M, et al. (2020) Enhancing the stability of adalimumab by engineering additional glycosylation motifs. International journal of biological macromolecules 158:189-196.
- Kayser V, et al. (2011) Evaluation of a Non-Arrhenius Model for Therapeutic Monoclonal Antibody Aggregation. Journal of Pharmaceutical Sciences 100(7):2526-2542.
- Wang W & Roberts CJ (2013) Non-Arrhenius protein aggregation. The AAPS journal 15(3):840-851.
- Henry KA (2018) Next-Generation DNA Sequencing of VH/VL Repertoires: A Primer and Guide to Applications in Single-Domain Antibody Discovery. Methods Mol Biol 1701:425-446.
- Hunter M, Yuan P, Vavilala D, & Fox M (2019) Optimization of Protein Expression in Mammalian Cells. Current Protocols in Protein Science 95(1):e77.
- Fliedl L, Grillari J, & Grillari-Voglauer R (2015) Human cell lines for the production of recombinant proteins: on the horizon. New Biotechnology 32(6):673-679.
- Lai T, Yang Y, & Ng SK (2013) Advances in Mammalian cell line development technologies for recombinant protein production. Pharmaceuticals (Basel, Switzerland) 6(5):579-603.
- Frenzel A, Hust M, & Schirrmann T (2013) Expression of Recombinant Antibodies. Frontiers in Immunology 4(217).
- Tran K, et al. (2018) Cell-free production of a therapeutic protein: Expression, purification, and characterization of recombinant streptokinase using a CHO lysate. Biotechnology and Bioengineering 115(1):92-102.
- Stech M, et al. (2017) Cell-free synthesis of functional antibodies using a coupled in vitro transcription-translation system based on CHO cell lysates. Scientific Reports 7(1):12030.
- Matsuda T, et al. (2018) Cell-free synthesis of functional antibody fragments to provide a structural basis for antibody–antigen interaction. PLOS ONE 13(2):e0193158.
- Li J, Wang H, Kwon Y-C, & Jewett MC (2017) Establishing a high yielding streptomyces-based cell-free protein synthesis system. Biotechnology and Bioengineering 114(6):1343-1353.
- Ulmer N, Vogg S, Müller-Späth T, & Morbidelli M (2019) Purification of Human Monoclonal Antibodies and Their Fragments. Human Monoclonal Antibodies: Methods and Protocols, ed Steinitz M (Springer New York, New York, NY), pp 163-188.
- Kelley BB, Greg; Lee, Ann (Downstream Processing of Monoclonal Antibodies: Current Practices and Future Opportunities. Process Scale Purification of Antibodies), pp 1-23.
- Sanford M (2014) Subcutaneous trastuzumab: a review of its use in HER2-positive breast cancer. Targeted Oncology 9(1):85-94.
- Jackisch C, et al. (2014) Subcutaneous versus intravenous formulation of trastuzumab for HER2-positive early breast cancer: updated results from the phase III HannaH study. Annals of Oncology 26(2):320-325.
- Reslan M & Kayser V (2018) Ionic liquids as biocompatible stabilizers of proteins. Biophysical Reviews 10(3):781-793.
- Crommelin DJA, Hawe A, & Jiskoot W (2019) Formulation of Biologics Including Biopharmaceutical Considerations. Pharmaceutical Biotechnology: Fundamentals and Applications, eds Crommelin DJA, Sindelar RD, & Meibohm B (Springer International Publishing, Cham), pp 83-103.
- Reslan M, Ranganathan V, MacFarlane D, & Kayser V (2018) Choline ionic liquid enhances the stability of Herceptin® (trastuzumab). Chemical Communications 54(75):10622-10625.
- Viola M, et al. (2018) Subcutaneous delivery of monoclonal antibodies: How do we get there? Journal of Controlled Release 286:301-314.
- Garidel P, Kuhn AB, Schäfer LV, Karow-Zwick AR, & Blech M (2017) High-concentration protein formulations: How high is high? European Journal of Pharmaceutics and Biopharmaceutics 119:353-360.
- Cruz E & Kayser V (2019) Synthesis and Enhanced Cellular Uptake In Vitro of Anti-HER2 Multifunctional Gold Nanoparticles. Cancers 11(6):870.
- Shi J, Kantoff PW, Wooster R, & Farokhzad OC (2016) Cancer nanomedicine: progress, challenges and opportunities. Nature Reviews Cancer 17:20.
- Lambert JM & Morris CQ (2017) Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Advances in Therapy 34(5):1015-1035.
- Dalziel M, Beers SA, Cragg MS, & Crispin M (2018) Through the barricades: overcoming the barriers to effective antibody-based cancer therapeutics. Glycobiology 28(9):697-712.
- Schweizer D, Serno T, & Goepferich A (2014) Controlled release of therapeutic antibody formats. European Journal of Pharmaceutics and Biopharmaceutics 88(2):291-309.



