 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kevin Pruitt | + 3170 word(s) | 3170 | 2020-11-10 10:53:41 | | | |
| 2 | Monica Sharma | Meta information modification | 3170 | 2020-11-12 21:59:47 | | | | |
| 3 | Peter Tang | -202 word(s) | 2968 | 2020-11-13 07:39:10 | | |
Video Upload Options
Wnt signaling has been implicated in a wide spectrum of important biological phenomena, where either a deficiency or overactivation of key effectors can lead to various human diseases. This review highlights historical and recent findings on key mediators of Wnt signaling and its association with various developmental diseases and tumorigenesis.
1. Introduction
Wnt signaling and its components have been implicated in a wide spectrum of important biological phenomena, where either a deficiency or overactivation of key effectors can lead to developmental disorders or impact cancer risk. The emergence of Wnt signaling, an integral mode of cell-to-cell communication, can be traced back to the early discovery of the first mammalian Wnt gene, Int1, discovered in 1982 [1]. Following the discovery of Int1, some of the most impactful discoveries were made which linked the Wnt pathway with developmental biology. Many of the genes regulated via the Wnt pathway, which were initially discovered to be important for development, later turned out to be oncogenes and tumor suppressors when studied in human cancer. Subsequently, some of the early genetic screens involving mutations in armadillo (β-catenin in vertebrates) and dishevelled (dsh in Drosophila) were similar to the Wingless mutants and thus shown to play an important role in segment polarity and embryonic development. Soon developmental assays, such as axis duplication assays in Xenopus, were shown to be excellent experimental model systems to characterize different components of the Wnt pathway. This led to classification of positive regulators of the Wnt pathway, for instance, wherein injection of murine Wnt1 mRNA into the embryo could induce axis duplication. Similarly, axis duplication was also induced by other components such as β-catenin, TCF, LEF, and GSK3β. In contrast, injection of Axin and APC mRNA resulted in complete loss of duplication, thereby identifying the negative regulators of the Wnt pathway. There was no major connection between the Wnt pathway and human cancer, until 1993 when a few research groups [2][3][4] reported the role of tumor suppressor APC and β-catenin in the Wnt pathway. Later, mutations in APC, β-catenin, and Axin were associated with genetic diseases such as familial adenomatous polyposis (FAP) in patients. These findings, for the first time, established a direct link between the Wnt pathway and human cancer.
Since the early discoveries, several genetic and biochemical studies have identified novel signaling components and provided deeper insights into the Wnt pathway. The known components of Wnt signaling include Wnt ligands, receptors and co-receptors (Frizzled and LRP), components of β-catenin destruction complex, and other transcriptional regulators within the nucleus. With recent advancements in structural biology and comprehensive genomic studies, it is clearly evident that the Wnt pathway is integral for developmental biology and plays a critical role in human cancer.
2. Overview of the Wnt Signaling Pathway
The Wnt pathway is one of the major signaling cascades that contributes to both normal development and pathophysiology. Herein, we will discuss the key components of the Wnt pathway and refer to some excellent reports which have laid the foundation and some recent findings that provide deeper insights into the complexities of this integral pathway.
The Wnt pathway is divided into two branches: β-catenin dependent (canonical) and independent (noncanonical), as outlined in Figure 1. The scope of Wnt signaling involvement in normal development and pathophysiology is daunting. With the 19 Wnt ligands, 10 Frizzled (FZD) receptors, and 3 Dishevelled (DVL) proteins participating in signaling, the amount of information relayed is enormous. Typically, in the canonical branch, Wnt proteins (secreted glycoproteins) bind to a seven-pass transmembrane receptor protein called FZD and low-density lipoprotein receptor-related protein 5/6 (LRP5/6) to activate the Wnt signaling pathway at the plasma membrane. In the absence of Wnts, β-catenin is degraded via a destruction complex consisting of tumor suppressors such as adenomatous polyposis coli (APC), Axin, glycogen synthase kinase-3β (GSK3β), and casein kinase 1 (CK1). This destruction complex induces phosphorylation of β-catenin on key serine and threonine residues (Ser33, Ser37, Ser 45, and Thr41) by CK1 and GSK3β, resulting in the ubiquitination by E3 ubiquitin ligase β-TrCP, and subsequent proteasomal degradation of β-catenin. Conversely, Wnt protein secretion results in activation of FZD and LRP5/6 receptors on the plasma membrane, permitting binding of DVL proteins. Sequential phosphorylation of cytoplasmic motifs on LRP5/6 receptors allows its interaction with Axin, resulting in destabilization of β-catenin destruction complex. Eventually, dephosphorylated β-catenin becomes stabilized and translocates into the nucleus to interact with transcription factor/lymphoid enhancer-binding factor (TCF/LEF) to initiate transcription of Wnt target genes. The β-catenin and TCF complex mediated transcriptional activation plays a crucial role in regulation of diverse cell behaviors, including cell fate, cell survival, proliferation, and stem cell renewal. In recent years, novel findings about the canonical Wnt pathway allowed the model to be refined and provided deeper insights into how this pathway is regulated. For instance, we now understand that proper production and secretion of Wnt ligands is a crucial step for Wnt pathway activation. An ER resident enzyme such as Porcupine, an acyl-transferase, is essential for the attachment of palmitoleic acid to Wnt ligands. This allows the lipid-modified Wnt ligands to bind to transmembrane protein Evi/WIs which are shuttled to plasma membrane for secretion [5]. Interestingly, a variety of mechanisms have been proposed for the short-range versus long-range release of Wnt ligands that may correspond to the role of Wnt in either development or cellular maintenance. For example, depending on the need, the levels and the range of Wnt signaling vary between the process of intestinal organoid and sperm maturation, suggesting that tissue-specific mechanisms may exist. Beyond the Wnt ligands, several other regulators have been demonstrated to play a crucial role in Wnt activation. For instance, R-spondin ligands serve as positive regulators and bind to leucine-rich repeat-containing G-protein-coupled receptor (Lgr4-6). In the absence of R-spondin, ZNRF3/RNF43, two homologues of E3 ubiquitin ligases, bind to FZD and target it for degradation. However, when R-spondin is present, it interacts with Lgr4-6, inhibiting the activity of ZNRF3/RNF43 and thereby allowing accumulation of FZD receptors on the plasma membrane. It is fascinating to see how Wnt target genes such as ZNRF3 and RNF43, function as negative feedback regulators in Lgr5-positive cells. Moreover, recent studies have highlighted the importance of R-spondin, Lgr5, and RNF43 in different cancers types, including colorectal cancer, which harbor inactivating mutations on RNF43 [6]. A surprising number of cytoplasmic Wnt regulators, including APC, Axin, and DVL, have been found to localize within the nucleus. APC and Axin, have been found to contain nuclear import and export sequences that direct them to shuttle in and out of the nucleus [7]. While the classical model asserts that Dishevelled functions in the cytosol, recent studies have shown that DVL proteins translocate into the nucleus via a regulatory post-translational acetylation switch in breast cancer cells [8][9]. Recent findings indicate that oncogenic Yes-associated protein (YAP) of the Hippo pathway paradoxically suppresses Wnt activity. The study reported that Wnt scaffolding protein Dishevelled (DVL) is responsible for cytosolic translocation of phosphorylated YAP [10]. This mechanistic study complements previous evidence in showing that YAP is a part of β-catenin destruction complex, thereby acting as a negative regulator of Wnt signaling. Recent findings have shown that alternative tyrosine-kinase related receptors (such as MET, FER, and FYN), and nonreceptor tyrosine kinases (such as SRC and ABL) could enhance of β-catenin and TCF complex mediated transcription by disrupting interaction of E-cadherin with β-catenin. Additionally, G-protein-coupled receptor signal transduction and environmental conditions such as hypoxia and high glucose levels could activate TCF-β-catenin signaling [11].
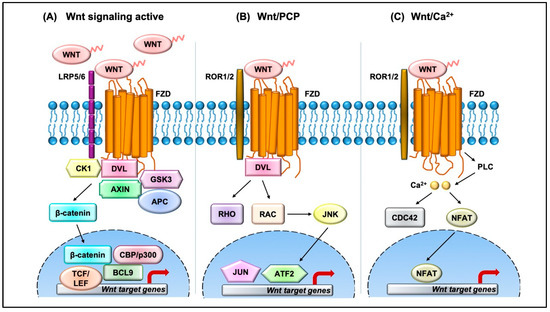
Figure 1. Overview of the Wnt signaling pathway. (A) In the canonical Wnt pathway, secreted Wnt ligands (usually Wnt3A and Wnt1) bind to Frizzled (FZD) receptors and LRP co-receptors. These receptors are then activated via CK1 and GSK3B mediated phosphorylation, which further recruits Dishevelled (DVL) to the plasma membrane and initiates activation. The DVL signalosome results in sequestration and inhibition of β-catenin destruction complex (Axin and APC) allowing stabilized β-catenin levels in the cytoplasm to increase. β-Catenin translocates into the nucleus where it forms an active complex with lymphoid enhancer factor (LEF), T-cell factor (TCF), and other histone-modifying co-activators such as CBP/p300 and BCL9, resulting in transcriptional activation (as represented by red arrow) of Wnt target genes which in turn causes an increase in cellular processes such as cell proliferation and differentiation and stem cell renewal. (B) In the Wnt/PCP pathway, Wnt ligands bind to FZD and co-receptors such as ROR1/2 and recruit DVL to the plasma membrane. DVL interacts with small GTPases such as RHO and RAC to further trigger activation of JNK. This results in activation of cytoskeletal rearrangements by transcriptional responses via JUN and ATF2 (activating transcription factor). (C) The Wnt/Ca2+ pathway is initiated by Wnt-FZD-ROR complex and G-protein-triggered phospholipase C (PLC) activity that results in intracellular Ca2+ influx. This further activates CDC42 and triggers calcium-dependent cell movement and polarity via various transcriptional responses (red arrow).
The noncanonical branch is independent of β-catenin and, even though less characterized, regulates more diverse cellular function such as cell organization and polarity, allowing it to be further classified into the planar cell polarity (PCP) pathway and Wnt/Ca2+ pathway. In the PCP pathway, Wnt ligands (usually Wnt 5a and Wnt 11) bind to panel of receptors including FZD and tyrosine kinase co-receptors such as ROR1/2 and Ryk. DVL relays noncanonical Wnt signals and interacts with Rac1 and DVL-associated activator of morphogenesis 1 (DAAM1). Rac1 activates downstream c-Jun kinases, while DAAM1 activates Rho to further activate Rho-associated kinase (ROCK), eventually regulating actin polymerization and cellular cytoskeletal arrangements. In the Wnt/Ca2+ pathway, the Wnt ligands bind to FZD which interacts with heterotrimeric G-proteins and DVL, resulting in activation of phospholipase C (PLC) and an intracellular increase in Ca2+ levels. This cascade results in downstream activation of downstream signaling proteins such as protein kinase C (PKC), calcineurin, and Ca2+/calmodulin-dependent protein kinase II (CaMKII) that regulate cell adhesion and cell migration. Wnt5a-mediated activation of the Wnt/Ca2+ branch antagonizes canonical Wnt/β-catenin signaling by phosphorylating TCF4 via Nemo-like kinase, thus preventing the binding of the β-catenin-TCF4 complex to DNA [12][13], suggesting a coordinated interplay between the different branches of the Wnt pathway.
Together, Wnt signals incorporated into the canonical and noncanonical pathways regulate complex normal cellular processes such as cell differentiation, development, tissue homeostasis, and wound healing; however, when the Wnt pathway is aberrantly regulated, it can be associated with developmental disorders, tumorigenesis, and other diseases.
3. Wnt Signaling in Human Diseases
Since Wnt signaling coordinates cell development processes and adult tissue homeostasis, it is certain that its deregulation can be linked with developmental disorders, cancer, and other diseases. Table 1 provides a list of some of the diseases associated with mutations in the Wnt signaling components. These include mutations in various Wnt ligands and components involved in both canonical and noncanonical branches of the Wnt pathway, shedding light on Wnt regulation in human development. For instance, increased expression of Wnt1 results in synaptic rearrangement in patients with schizophrenia. Tetra-amelia, a rare human genetic disease characterized by absence of four limbs, has been proposed to be caused by a nonsense mutation in the Wnt3 gene. Similarly, mutation in the Wnt4 gene is linked to intersex phenotype, Mullerian-duct regression, and kidney developmental defects. Other development disorders such as Fuhrmann syndrome, Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome, and Santos Syndrome are linked with loss-of-function mutation in Wnt7A gene. Mutations in Wnt5B and Wnt10B may be linked to type II diabetes and obesity, respectively. Aberrant Wnt signaling may play a role in precancerous conditions such as hypohidrotic ectodermal dysplasia and odonto-onycho-dermal dysplasia as a result of Wnt10A mutations. Mutations in either Fzd4 or LRP5 genes are associated with familial exudative vitreoretinopathy (FEVR). Moreover, heterozygous mutations in Fzd2 are implicated to be involved with cardiovascular diseases and a rare skeletal disorder (known as omodysplasia) characterized by severe limb shortening and facial dysmorphism. The Wnt pathway is also involved in controlling bone mass, a discovery that stemmed from analysis of LRP5 mutation in patients with osteoporosis-pseudoglioma syndrome (OPPG), a disorder characterized by low bone-mass density. Conversely, patients who harbor LRP5 gain-of-function mutations experience high bone mass disease. Moreover, LRP6 mutations are also linked with neuronal and metabolic disorders such as Alzheimer's and coronary artery disease. The central mediators of the Wnt pathway, Dishevelled genes, are also implicated in human disease. Early discoveries suggested that DVL genes were imperative for segment polarity in wing hair of Drosophila and Xenopus embryos [14]. Moreover, DVL knockout mice were shown to exhibit abnormal social interaction in nest building, home cage huddling, neural tube closure, and cardiovascular malformations [15][16][17]. In humans, mutations in DVL are associated with severe disorders such as Schwartz-Jampel syndrome, Charcot-Marie-Tooth disease type 2A, DiGeorge syndrome, and Hirschsprung's disease [18][19]. Recent promising evidence suggests that frameshift mutations on C-terminal tail of all three DVL genes can cause Robinow syndrome, characterized by skeletal abnormalities [20][21][22][23][24]. To summarize, DVL plays an important role in development, and mutations in DVL genes can lead to severe phenotypic defects. Recently, components of Wnt signaling such as DVL were reported to play an instrumental role in developing neural circuits and adult brain function and cause neurodevelopmental disorders such as autism spectrum disorders (ASDs) and intellectual disability (ID) [25].
Table 1. Human diseases associated with dysregulation in Wnt signaling components.
|
Gene |
Dysfunction |
Associated Disease |
References |
|
Wnt 1 |
Gain of function |
Schizophrenia |
[26] |
|
Wnt 3 |
Loss of function |
Tetra-amelia |
[27] |
|
Wnt 4 |
Gain and loss of function |
Intersex phenotype (GOF), kidney development, Mullerian duct regression and virilization (LOF) |
|
|
Wnt 5B |
Gain of function |
Type II diabetes, breast tumorigenesis |
|
|
Wnt 7A |
Loss of function |
Fuhrmann syndrome, Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome, Santos syndrome |
|
|
Wnt 10A |
Loss of function |
Hypohidrotic ectodermal dysplasia, odonto-onycho-dermal dysplasia |
|
|
Wnt 10B |
Loss of function |
Obesity, reduced bone mass |
|
|
Fzd 2 |
Heterozygous mutations |
Omodysplasia, cardiovascular disease |
|
|
Fzd 4 |
Loss of function |
Familial exudative vitreoretinopathy (FEVR) |
|
|
LRP5 |
Gain and loss of function |
High bone mass (GOF), osteoporosis-pseudoglioma syndrome (LOF), familial exudative vitreoretinopathy (FEVR) |
|
|
LRP6 |
Gain and loss of function |
Alzheimer's disease (LOF), coronary artery disease (LOF), osteoarthritis (GOF) |
|
|
DVL1 |
Gain and loss of function |
Robinow syndrome (frameshift mutation), Schwartz–Jampel syndrome, Charcot–Marie–Tooth disease type 2A and DiGeorge syndrome, myocardial infarction, Hirschsprung's disease (GOF), autism spectrum disorders |
|
|
DVL2 |
Loss of function |
Robinow syndrome, defect in cardiac outflow tract formation |
|
|
DVL3 |
Gain and loss of function |
Hirschsprung's disease (GOF), autism spectrum disorders |
|
|
Axin 1 |
Loss of function |
Caudal duplication anomalies, gastrointestinal cancers, colorectal cancer, hepatocellular carcinomas, sporadic medulloblastoma, breast cancer |
|
|
Axin 2 |
Loss of function |
Congenital heart defects, familial tooth agenesis, predisposition to multiple cancers including hepatocellular carcinoma and colorectal, prostate, ovarian, and lung cancers |
|
|
APC |
Loss of function |
Familial adenomatous polyposis (FAP), colon cancer |
|
|
GSK3β |
Altered activity |
Alzheimer's disease, diabetes, schizophrenia, bipolar disorder, and cancer |
|
|
β-Catenin |
Gain and loss of function |
Cancer (GOF), Alzheimer's disease (LOF) |
|
|
TCF4 |
Transcript variants |
Pitt–Hopkins syndrome, schizophrenia, Fuchs' endothelial corneal dystrophy, primary sclerosing cholangitis, type II diabetes |
|
|
JNK |
Altered activity |
Obesity, type II diabetes, nonalcoholic fatty liver disease (NAFLD) |
[77] |
|
Rho/Rac |
Altered activity |
Alzheimer's disease, cardiovascular disease, leukocyte adhesion deficiency (LAD) |
|
|
TIAM1 |
Altered activity |
Cardiovascular disease and cancer |
|
|
NFAT |
Loss of function |
Down's syndrome |
[84] |
Deregulation of Wnt/β-catenin signaling also predisposes patients to multiple cancer types such as colorectal, hepatocellular carcinoma, ovarian, and lung cancer. In particular, loss-of-function mutations in APC, Axin1, and Axin, have been well documented in the cancer types mentioned above. Conversely, gain-of-function mutation of β-catenin has been established in colorectal cancer with wild-type APC. In contrast, attenuated β-catenin signaling may be implicated in the development of Alzheimer's disease, a neurodegenerative disease characterized by deposits of the amyloid β-peptide and selective death of neurons. Recently, increasing evidence has shown that GSK-3β may be a key link between diabetes mellitus (DM) and Alzheimer's disease (AD), where GSK-3β controls glycogen synthesis, thereby regulating blood glucose [70]. Genome-wide association studies have illustrated the role of TCF4 in these seemingly diverse disorders. TCF4 gene has been strongly implicated in type II diabetes; however, recent data suggest that TCF4 is also an important regulator of neurodevelopment disorders such as schizophrenia, Fuchs' endothelial corneal dystrophy, and primary sclerosing cholangitis. However, rare TCF4 mutations causing Pitt–Hopkins syndrome, a disorder characterized by intellectual disability and developmental delay, have also been described in patients with other neurodevelopmental disorders [76].
Interestingly, since components of noncanonical branch also play a central role in the cell processes and stress response, mutations in JNK, Rho/Rac, TIAM1, and NFAT have been associated with various disorders, including metabolic, neurodegenerative, and cardiovascular diseases. JNK is one of the most investigated signal transducers, and emerging evidence suggests that different isoforms of JNK (JNK1 and JNK2) may promote the development of obesity to insulin resistance in a cell-specific manner, NAFLD, and type II diabetes. This has led to development of isoform-specific JNK inhibitors with specific tissue distribution as possible drug targets for the treatment of type II diabetes [77]. Moreover, recent evidence suggests that excessive activity of the RhoA/Rac-kinase pathway, widely known to play important roles in many cellular functions, promotes the development of AD pathogenesis and cardiovascular diseases [78][79]. Recently, mutations in the hematopoietic specific GTPase, RAC2, have been found to cause a human disease, a severe phagocytic immunodeficiency characterized by life-threatening infections in infancy. Interestingly, the phenotype was predicted by a mouse knockout of Rac2 and resembles leukocyte adhesion deficiency (LAD) [80]. The activity of T-cell lymphoma invasion and metastasis 1 (TIAM1), a Rac guanine nucleotide exchange factor (GEF), crucial for cell adhesion and migration, has been demonstrated to be upregulated in some cancers [81][82]. Additionally, TIAM1 has also shown to play a pivotal role in cardiac hypertrophy associated with heart failure [85]. Mathematical models and in vivo studies predict that restriction of nuclear occupancy of NFAT transcription factors, resulting in inactivation of NFAT target genes, may cause Down's syndrome (chromosomal trisomy), further causing neurological, skeletal, cardiovascular, and immunological defects [84]. More generally, these findings suggest that the destabilization of important regulatory pathways such as Wnt signaling can underlie human diseases.
References
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684.
- Su, L.K.; Vogelstein, B.; Kinzler, K.W. Association of the APC tumor suppressor protein with catenins. Science 1993, 262, 1734–1737.
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665.
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398.
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810.
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473.
- Willert, K. Wnt signaling: Is the party in the nucleus? Genes Dev. 2006, 20, 1394–1404.
- Castro-Piedras, I.; Sharma, M.; Bakker, M.D.; Molehin, D.; Martinez, E.; Vartak, D.; Pruitt, W.M.; Deitrick, J.; Almodovar, S.; Pruitt, K. DVL1 and DVL3 differentially localize to CYP19A1 promoters and regulate aromatase mRNA in breast cancer cells. Oncotarget 2018, 9, 35639–35654.
- Sharma, M.; Molehin, D.; Castro-Piedras, I.; Martinez, E.G.; Pruitt, K. Acetylation of conserved DVL-1 lysines regulates its nuclear translocation and binding to gene promoters in triple-negative breast cancer. Sci. Rep. 2019, 9, 16257.
- Lee, Y.; Kim, N.H.; Cho, E.S.; Yang, J.H.; Cha, Y.H.; Kang, H.E.; Yun, J.S.; Cho, S.B.; Lee, S.-H.; Paclikova, P.; et al. Dishevelled has a YAP nuclear export function in a tumor suppressor context-dependent manner. Nat. Commun. 2018, 9, 1–16.
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532.
- Ishitani, T.; Ninomiya-Tsuji, J.; Matsumoto, K. Regulation of lymphoid enhancer factor 1/T-cell factor by mitogen-activated protein kinase-related Nemo-like kinase-dependent phosphorylation in Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003, 23, 1379–1389.
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310.
- Wallingford, J.B.; Habas, R. The developmental biology of Dishevelled: An enigmatic protein governingcell fate and cell polarity. Development 2005, 132, 4421–4436.
- Lijam, N.; Paylor, R.; McDonald, M.P.; Crawley, J.N.; Deng, C.-X.; Herrup, K.; Stevens, K.; Maccaferri, G.; McBain, C.J.; Sussman, D.J.; et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell 1997, 90, 895–905.
- Van Gijn, M.; Daemen, M.J.; Smits, J.F.; Blankesteijn, W.M. The wnt-frizzled cascade in cardiovascular disease. Cardiovasc. Res. 2002, 55, 16–24.
- Hamblet, N.S.; Lijam, N.; Ruiz-Lozano, P.; Wang, J.; Yang, Y.; Luo, Z.; Mei, L.; Chien, K.R.; Sussman, D.J.; Wynshaw-Boris, A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development 2002, 129, 5827–5838.
- Pizzuti, A.; Novelli, G.; Mari, A.; Ratti, A.; Colosimo, A.; Amati, F.; Penso, D.; Sangiuolo, F.; Calabrese, G.; Palka, G.; et al. Human homologue sequences to the Drosophila dishevelled segment-polarity gene are deleted in the DiGeorge syndrome. Am. J. Hum. Genet. 1996, 58, 722–729.
- Chen, N.; Mi, J.; Wu, M.; Wang, W.; Gao, H. Expression of dishevelled gene in Hirschsprung’s disease. Int. J. Clin. Exp. Pathol. 2013, 6, 1791–1798.
- Person, A.D.; Beiraghi, S.; Sieben, C.M.; Hermanson, S.; Neumann, A.N.; Robu, M.E.; Schleiffarth, J.R., Jr.; Van Bokhoven, H.; Hoogeboom, J.M.; et al. WNT5Amutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn. 2010, 239, 327–337.
- White, J.; Mazzeu, J.F.; Hoischen, A.; Jhangiani, S.N.; Gambin, T.; Alcino, M.C.; Penney, S.; Saraiva, J.M.; Hove, H.; Skovby, F.; et al. DVL1 Frameshift Mutations Clustering in the Penultimate Exon Cause Autosomal-Dominant Robinow Syndrome. Am. J. Hum. Genet. 2015, 96, 612–622.
- Bunn, K.J.; Daniel, P.; Roesken, H.S.; O’Neill, A.C.; Cameron-Christie, S.R.; Morgan, T.; Brunner, H.G.; Lai, A.; Kunst, H.P.M.; Markie, D.M.; et al. Mutations in DVL1 Cause an Osteosclerotic Form of Robinow Syndrome. Am. J. Hum. Genet. 2015, 96, 623–630.
- Mansour, T.; Lucot, K.L.; Konopelski, S.E.; Dickinson, P.J.; Sturges, B.K.; Vernau, K.L.; Choi, S.; Stern, J.A.; Thomasy, S.; Döring, S.; et al. Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related screw tail dog breeds. PLoS Genet. 2018, 14, e1007850.
- White, J.J.; Mazzeu, J.F.; Hoischen, A.; Bayram, Y.; Withers, M.; Gezdirici, A.; Kimonis, V.; Steehouwer, M.; Jhangiani, S.N.; Muzny, D.M.; et al. DVL3 Alleles Resulting in a −1 Frameshift of the Last Exon Mediate Autosomal-Dominant Robinow Syndrome. Am. J. Hum. Genet. 2016, 98, 553–561.
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45.
- Miyaoka, T.; Seno, H.; Ishino, H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999, 38, 1–6.
- Niemann, S.; Zhao, C.; Pascu, F.; Stahl, U.; Aulepp, U.; Niswander, L.; Weber, J.L.; Müller, U. Homozygous WNT3 Mutation Causes Tetra-Amelia in a Large Consanguineous Family. Am. J. Hum. Genet. 2004, 74, 558–563.
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 Mutation Associated with Müllerian-Duct Regression and Virilization in a 46, XX Woman. N. Engl. J. Med. 2004, 351, 792–798.
- Jordan, B.K.; Mohammed, M.; Ching, S.T.; Délot, E.; Chen, X.-N.; Dewing, P.; Swain, A.; Rao, P.N.; Elejalde, B.R.; Vilain, E. Up-Regulation of WNT-4 Signaling and Dosage-Sensitive Sex Reversal in Humans. Am. J. Hum. Genet. 2001, 68, 1102–1109.
- Perantoni, A.O. Renal development: perspectives on a Wnt-dependent process. Semin. Cell Dev. Biol. 2003, 14, 201–208.
- Park, J.S.; Valerius, M.T.; McMahon, A.P. Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 2007, 134, 2533–2539.
- Kanazawa, A.; Tsukada, S.; Sekine, A.; Tsunoda, T.; Takahashi, A.; Kashiwagi, A.; Tanaka, Y.; Babazono, T.; Matsuda, M.; Kaku, K.; et al. Association of the Gene Encoding Wingless-Type Mammary Tumor Virus Integration-Site Family Member 5B (WNT5B) with Type 2 Diabetes. Am. J. Hum. Genet. 2004, 75, 832–843.
- Jiang, S.; Zhang, M.; Zhang, Y.; Zhou, W.; Zhu, T.; Ruan, Q.; Chen, H.; Fang, J.; Zhou, F.; Sun, J.; et al. WNT5B governs the phenotype of basal-like breast cancer by activating WNT signaling. Cell Commun. Signal. 2019, 17, 1–19.
- Alves, L.U.; Santos, S.; Musso, C.M.; Ezquina, S.A.; Opitz, J.M.; Kok, F.; Otto, P.; Mingroni-Netto, R.C. Santos syndrome is caused by mutation in the WNT7A gene. J. Hum. Genet. 2017, 62, 1073–1078.
- Woods, C.G.; Stricker, S.; Seemann, P.; Stern, R.; Cox, J.; Sherridan, E.; Roberts, E.; Springell, K.; Scott, S.; Karbani, G.; et al. Mutations in WNT7A Cause a Range of Limb Malformations, Including Fuhrmann Syndrome and Al-Awadi/Raas-Rothschild/Schinzel Phocomelia Syndrome. Am. J. Hum. Genet. 2006, 79, 402–408.
- Xu, M.; Horrell, J.; Snitow, M.; Cui, J.; Gochnauer, H.; Syrett, C.M.; Kallish, S.; Seykora, J.T.; Liu, F.; Gaillard, D.; et al. WNT10A mutation causes ectodermal dysplasia by impairing progenitor cell proliferation and KLF4-mediated differentiation. Nat. Commun. 2017, 8, 15397.
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; De Mazancourt, P.; Megarbane, A. Mutation in WNT10A Is Associated with an Autosomal Recessive Ectodermal Dysplasia: The Odonto-onycho-dermal Dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828.
- Christodoulides, C.; Scarda, A.; Granzotto, M.; Milan, G.; Nora, E.D.; Keogh, J.; De Pergola, G.; Stirling, H.; Pannacciulli, N.; Sethi, J.K.; et al. WNT10B mutations in human obesity. Diabetologia 2006, 49, 678–684.
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329.
- Türkmen, S.; Spielmann, M.; Güneş, N.; Knaus, A.; Flöttmann, R.; Mundlos, S.; Tüysüz, B. A Novel de novo FZD2 Mutation in a Patient with Autosomal Dominant Omodysplasia. Mol. Syndr. 2017, 8, 318–324.
- Robitaille, J.; MacDonald, M.L.; Kaykas, A.; Sheldahl, L.C.; Zeisler, J.; Dubé, M.-P.; Zhang, L.-H.; Singaraja, R.R.; Guernsey, D.L.; Zheng, B.; et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 2002, 32, 326–330.
- Kondo, H.; Hayashi, H.; Oshima, K.; Tahira, T.; Hayashi, K. Frizzled 4 gene (FZD4) mutations in patients with familial exudative vitreoretinopathy with variable expressivity. Br. J. Ophthalmol. 2003, 87, 1291–1295.
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High Bone Density Due to a Mutation in LDL-Receptor–Related Protein 5. N. Engl. J. Med. 2002, 346, 1513–1521.
- Little, R.D.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.-C.; Eustace, B.; Lappe, M.M.; Spitzer, L.; Zweier, S.; Braunschweiger, K.; et al. A Mutation in the LDL Receptor–Related Protein 5 Gene Results in the Autosomal Dominant High–Bone-Mass Trait. Am. J. Hum. Genet. 2002, 70, 11–19.
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL Receptor-Related Protein 5 (LRP5) Affects Bone Accrual and Eye Development. Cell 2001, 107, 513–523.
- Korvala, J.; Jüppner, H.; Mäkitie, O.; Sochett, E.; Schnabel, D.; Mora, S.; Bartels, C.F.; Warman, M.L.; Deraska, D.; Cole, W.G.; et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet. 2012, 13, 26.
- Pefkianaki, M.; Hasanreisoglu, M.; Suchy, S.F.; Shields, C.L. Familial Exudative Vitreoretinopathy With a NovelLRP5Mutation. J. Pediatr. Ophthalmol. Strabismus 2016, 53, e39–e42.
- Joiner, D.M.; Ke, J.; Zhong, Z.; Xu, H.E.; Williams, B.O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 2013, 24, 31–39.
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; De-Vrieze, F.W.; Avila, M.E.; Major, M.B.; Myers, A.; Sáez, K.; Henríquez, J.P.; Zhao, A.; et al. Common genetic variation within the Low-Density Lipoprotein Receptor-Related Protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 9434–9439.
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, M.-A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; Lifton, R.P. LRP6 Mutation in a Family with Early Coronary Disease and Metabolic Risk Factors. Science 2007, 315, 1278–1282.
- Velasco, J.; Zarrabeitia, M.T.; Prieto, J.R.; Pérez-Castrillón, J.L.; Perez-Aguilar, M.D.; Perez-Nuñez, M.I.; Sanudo, C.; Hernandez-Elena, J.; Calvo, I.; Ortiz, F.; et al. Wnt pathway genes in osteoporosis and osteoarthritis: Differential expression and genetic association study. Osteoporos. Int. 2009, 21, 109–118.
- Oates, N.A.; Van Vliet, J.; Duffy, D.L.; Kroes, H.Y.; Martin, N.G.; Boomsma, D.I.; Campbell, M.; Coulthard, M.; Whitelaw, E.; Chong, S. Increased DNA Methylation at the AXIN1 Gene in a Monozygotic Twin from a Pair Discordant for a Caudal Duplication Anomaly. Am. J. Hum. Genet. 2006, 79, 155–162.
- Kroes, H.Y.; Takahashi, M.; Zijlstra, R.; Baert, J.; Kooi, K.; Hofstra, R.M.W.; Van Essen, A.J. Two cases of the caudal duplication anomaly including a discordant monozygotic twin. Am. J. Med Genet. 2002, 112, 390–393.
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014, 355, 1–8.
- Picco, G.; Petti, C.; Centonze, A.; Torchiaro, E.; Crisafulli, G.; Novara, L.; Acquaviva, A.; Bardelli, A.; Medico, E. Loss of AXIN1 drives acquired resistance to WNT pathway blockade in colorectal cancer cells carrying RSPO3 fusions. EMBO Mol. Med. 2017, 9, 293–303.
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250.
- Dahmen, R.; Koch, A.; Denkhaus, D.; Tonn, J.C.; Sörensen, N.; Berthold, F.; Behrens, J.; Birchmeier, W.; Wiestler, O.D.; Pietsch, T. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001, 61, 7039–7043.
- Zhang, X.; Farrell, A.S.; Daniel, C.J.; Arnold, H.; Scanlan, C.; Laraway, B.; Janghorban, M.; Lum, L.; Chen, D.; Troxell, M.; et al. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proc. Natl. Acad. Sci. USA 2012, 109, 2790–2795.
- Zhu, M.-J.; Ma, X.-Y.; Ding, P.-C.; Tang, H.-F.; Peng, R.; Lu, L.; Li, P.-Q.; Qiao, B.; Yang, X.-Y.; Zheng, Y.-F.; et al. Novel mutations of AXIN2 identified in a Chinese Congenital Heart Disease Cohort. J. Hum. Genet. 2019, 64, 427–435.
- Lammi, L.; Arte, S.; Somer, M.; Järvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 Cause Familial Tooth Agenesis and Predispose to Colorectal Cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050.
- Callahan, N.; Modesto, A.; Meira, R.; Seymen, F.; Patir, A.; Vieira, A. Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch. Oral Biol. 2009, 54, 45–49.
- Hlouskova, A.; Bielik, P.; Bonczek, O.; Balcar, V.J.; Sery, O. Mutations in AXIN2 gene as a risk factor for tooth agenesis and cancer: A review. Neurol. Lett. 2017, 38, 131–137.
- Otero, L.; Lacunza, E.; Vasquez, V.; Arbelaez, V.; Cardier, F.; González, F. Variations in AXIN2 predict risk and prognosis of colorectal cancer. BDJ Open 2019, 5, 1–6.
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients.Science 1991, 253, 665–669.
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46.
- Egomez-Sintes, R.; Hernández, F.; Lucas, J.J.; Avila, J. GSK-3 mouse models to study neuronal apoptosis and neurodegeneration. Front. Mol. Neurosci. 2011, 4, 45.
- Kozlovsky, N.; Belmaker, R.H.; Agam, G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur. Neuropsychopharmacol. 2002, 12, 13–25.
- Li, X.; Liu, M.; Cai, Z.; Wang, G.; Li, X. Regulation of glycogen synthase kinase-3 during bipolar mania treatment. Bipolar Disord. 2010, 12, 741–752.
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3beta is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372.
- Zhang, Y.; Huang, N.-Q.; Yan, F.; Jin, H.; Zhou, S.-Y.; Shi, J.-S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65.
- Korinek, V.; Barker, N.; Morin, P.J.; Van Wichen, D.; De Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/-colon carcinoma. Science 1997, 275, 1784–1787.
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-Catenin-Tcf Signaling in Colon Cancer by Mutations in beta-Catenin or APC. Science 1997, 275, 1787–1790.
- He, P.; Shen, Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J Neurosci. 2009, 29, 6545–6557.
- Tamberg, L.; Sepp, M.; Timmusk, T.; Palgi, M. Introducing Pitt-Hopkins syndrome-associated mutations of TCF4 to Drosophila daughterless. Biol. Open 2015, 4, 1762–1771.
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.; José, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607.
- Forrest, M.P.; Hill, M.J.; Quantock, A.J.; Martin-Rendon, E.; Blake, D.J. The emerging roles of TCF4 in disease and development. Trends Mol. Med. 2014, 20, 322–331.
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184.
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 1–10.
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366.
- Pai, S.-Y.; Kim, C.; Williams, D.A. Rac GTPases in Human Diseases. Dis. Markers 2010, 29, 177–187.
- Contini, A.; Ferri, N.; Bucci, R.; Lupo, M.G.; Erba, E.; Gelmi, M.L.; Pellegrino, S. Peptide modulators of Rac1/Tiam1 protein-protein interaction: An alternative approach for cardiovascular diseases. Pept. Sci. 2018, 110, e23089.
- Izumi, D.; Toden, S.; Ureta, E.; Ishimoto, T.; Baba, H.; Goel, A. TIAM1 promotes chemoresistance and tumor invasiveness in colorectal cancer. Cell Death Dis. 2019, 10, 1–12.
- Saxena, M.; Dykes, S.S.; Malyarchuk, S.; Wang, A.E.; Cardelli, J.A.; Pruitt, K.M. The sirtuins promote Dishevelled-1 scaffolding of TIAM1, Rac activation and cell migration. Oncogene 2013, 34, 188–198.
- Arron, J.R., Winslow, M.M., Polleri, A., Chang, C.P., Wu, H., Gao, X., Neilson, J.R., Chen, L., Heit, J.J., Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600.
- Ferri, N.; Contini, A.; Bernini, S.K.; Corsini, A. Role of Small GTPase Protein Rac1 in Cardiovascular Diseases: Development of new selective pharmacological inhibitors. J. Cardiovasc. Pharmacol. 2013, 62, 425–435.


