 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yogeswaran Lokanathan | + 2575 word(s) | 2575 | 2020-11-02 09:12:10 | | | |
| 2 | Lily Guo | -40 word(s) | 2535 | 2020-11-06 06:46:13 | | |
Video Upload Options
Spinal cord injury (SCI) is a destructive neurological and pathological state that causes major motor, sensory and autonomic dysfunctions. Its pathophysiology comprises acute and chronic phases and incorporates a cascade of destructive events such as ischemia, oxidative stress, inflammatory events, apoptotic pathways and locomotor dysfunctions. This review aims to promote the understanding of SCI pathophysiology, interrelated or interlinked multimolecular interactions and various methods of neuronal recovery i.e., neuroprotective, immunomodulatory and neuro-regenerative pathways and relevant approaches.
1. Introduction
Spinal cord injury (SCI) is a devastating neurological state producing physical dependency, morbidity, psychological stress and financial burden. For the last 30 years, its global prevalence has increased from 236 to 1298 cases per million populations. The estimated global rate of SCI falls between 250,000 and 500,000 individuals every year[1]. The total lifetime costs for each patient with SCI exceed 3 million dollars, and the calculated annual economic burden is almost 2.67 billion dollars in Canada[2]. Available treatments are limited and only provide supportive relief to patients with lifetime disability[1]. Heterogeneous factors such as complex characteristics, abundant inconsistencies and complex pathophysiologic consequences post-SCI are the major reasons for poor understanding and failure of SCI treatment. Hip joint subluxation caused by SCI is challenging to overcome and causes lower leg paralysis[3]. SCI is also associated with autonomic dysreflexia (AD) occurring in 48%–60% of cases at above thoracic 6th vertebral level (T 6) and involving a sudden onset of excessively high blood pressure[4]. Understanding pathophysiology, phases and various wound recovery mechanisms associated with SCI is essential for the development of appropriate recovery treatments[5]. Normal spinal cord physiology involves interactions among many cell types such as astrocytes, neurons, microglia and oligodendrocytes. After a spinal injury, these multicellular interactions are interrupted and disorganised, leading to an impaired spinal recovery[5]. Various animal studies showed that the administration of current SCI treatments such as drugs, neuronal implants and stem cells induced the following improvements: (i) decrease neuro-inflammation, (ii) promote axonal growth, (iii) enhance myelination and (iv) reduce cavity size[2]. However, the current treatment strategies can aid for only a short duration and fail to completely overcome the detrimental effects of SCI. Therefore, knowledge on fundamental SCI pathophysiology and event sequences during and post-injury is beneficial in designing a suitable intervention for SCI[5]. Despite numerous studies and availability of various regenerative treatment strategies, post-SCI recovery remains controversial, and scientists are still exploring methods that could prevent or reverse the devastating outcomes of SCI[2].
2. SCI Phases
2.1. Primary Injury
Acute SCI commonly occurs due to sudden trauma to the spine and results in fractures and vertebrae dislocation. The initial stage immediately after the injury is known as primary injury[2][4] (Figure 1a) with features of bone fragments and spinal ligament tearing. SCI is accomplished in two phases: the first phase includes the destruction of neural parenchyma, disruption of axonal network, haemorrhage and disruption of glial membrane (Figure 1a). The main determinants for SCI severity are the extent of initial destruction and duration of spinal cord compression. A cascade of events associated with secondary injury is activated by the onset of biochemical, mechanical and physiological changes within neural tissues[6]. Although clinical manifestation suggests complete functional loss, few segments remain connected by some axons during primary SCI phase, thus reflecting incomplete and partial injury state[6][7].
2.2. Secondary Injury
The primary injury triggers secondary injury which produces further chemical and mechanical damage to spinal tissues, leads to neuronal excitotoxicity because of high calcium accumulation within cells and increases reactive oxygen concentrations and glutamate levels. These incidences damage underlying nucleic acid, proteins and phospholipids and result in neurological dysfunction[7]. The secondary injury phase reflects multi-featured pathological processes following the primary injury phase and lasts for several weeks (Figure 1b). Clinical manifestation of secondary injury includes increased cell permeability, apoptotic signalling, ischemia, vascular damage, oedema, excitotoxicity, ionic deregulation, inflammation, lipid peroxidation, free radical formation, demyelination, Wallerian degeneration, fibroglial scar and cyst formation as shown in Figure 2[7]. Disruption of blood vessels causes haemorrhage in spinal tissues, followed by invasion of monocytes, neutrophils, T and B lymphocytic cells and macrophages to spinal tissues. This phenomenon is also associated with the release of inflammatory cytokines such as interleukin (IL)-1a, IL-1b, IL-6 and tumour necrosis factor (TNF)-α after 6–12 h post-injury. The penetration of immune cells and inflammatory cytokines promotes the inflammation of neurons [8].
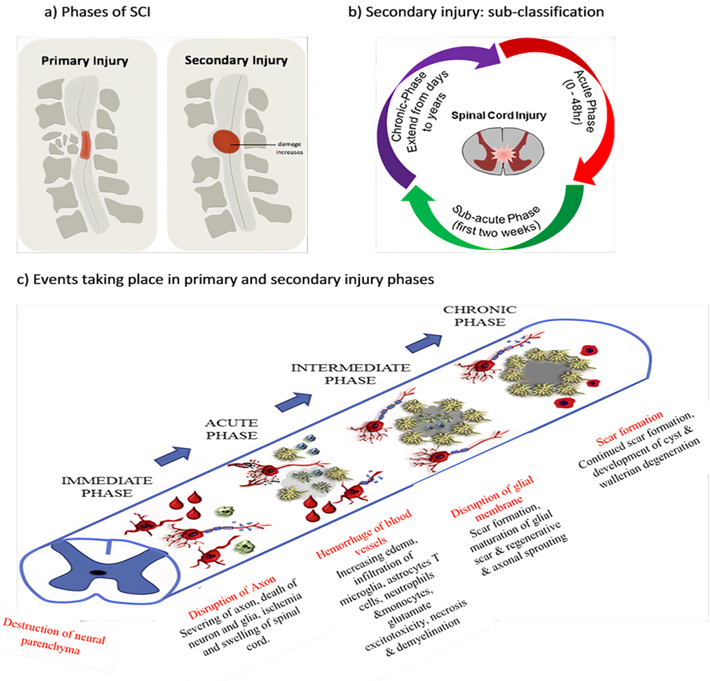
Figure 1. Spinal cord injury (SCI) (a) phases of SCI, (b) sub-classification of secondary injury depending on duration of injury and (c) pathophysiological events according to SCI phases.
The secondary injury is categorised into three phases: acute, sub-acute and chronic injury (Figure 1b). Following the primary injury phase, the initiation of acute secondary injury phase begins is manifested through clinical features such as vascular damage, ionic imbalance, excitotoxicity, free radical production, increased calcium influx, lipid peroxidation, inflammation, oedema and necrosis[9]. If the acute secondary injury phase persists, then the sub-acute secondary injury phase begins and is manifested by features such as neuronal apoptosis, axonal demyelination, Wallerian degeneration, axonal remodelling and glial scar formation[9] as shown in Figure 2. Sub-acute secondary injury leads to the chronic secondary injury phase of SCI as characterised by the formation of cystic cavity, axonal dieback, and maturation of glial scar[10].
3. Pathophysiology of SCI
SCI pathophysiology comprises interrelated events, each serving as the facilitator for the other. In some instances, multiple events occur simultaneously and cause complicated attributes, thus rendering this illness difficult to treat. SCI can be represented as a cascade of different interrelated events (Figure 2).
The most vulnerable clinical manifestation immediately after injury is the interruption of spinal cord vascular supply and hypotension/hypo-perfusion, producing hypovolemia, neurogenic shock and bradycardia. These signs occur because of extensive bleeding and neurogenic shock leading to spinal cord ischemia. The rupture of small blood vessels and capillaries promotes the extravasation of leukocytes and red blood cells (RBCs). These extravasations of immune cells at the injury site exert pressure on the injured spinal tissues and further disrupt the blood flow, thus producing vasospasm[9]. This state continues up to 24 h. Occurrence of vascular ischemia, hypovolemia and hyper-perfusion eventually leads to cell death and tissue destruction[9][10].
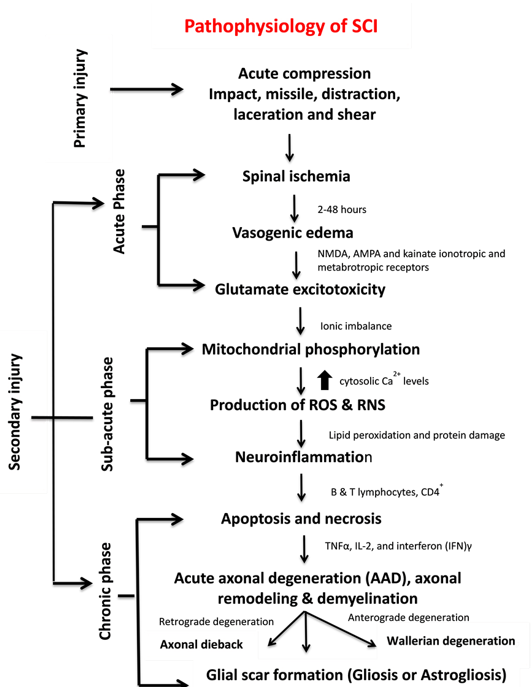
Figure 2. Pathophysiology, clinical manifestations, and phases of SCI.
Spinal cord ischemia causes cytotoxic, ionic and vasogenic oedemas. In normal physiology, the influx of Na+ occurs due to the passive influx of Cl− through chloride channels. Consequently, water molecules influx through aquaporin water channels. During a pathophysiological state, the balance between solute and water influx at the intracellular compartment is disturbed, thereby causing cell swelling and loss of cytoskeletal integrity and promoting cell death[11]. Ionic oedema occurs due to the increased permeability of the blood–spinal cord barrier that increases trans-endothelial ion transport and causes the loss of ions and water from the interstitial space[12]. Endothelial injury and inflammation subsequently increase the pore size and thus allow large plasma-derived molecules to pass through the cell membrane, resulting in vasogenic oedema[12]. This acute secondary injury phase continues from 2 h to 48 h. Continuous haemorrhage, oedema and inflammatory stage lead to substantial necrosis indicated by the increased concentration of specific inflammatory and the presence of structural biomarkers, e.g. glial fibrillary acidic protein (GFAP) or IL-6 in cerebrospinal fluid (CSF)[6]. These processes provoke free radical formation, glutamate-mediated excitotoxicity and neurotoxicity[12] (Figure 1c).
Glutamate is an excitatory neurotransmitter that is released in the central nervous system (CNS) and interacts with N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate ionotropic and metabotropic receptors[12] (Figure 2). The activation of glutamate receptors during SCI greatly increases glutamate concentrations and produces persistent excitotoxicity and cell death[12]. Abnormal increases in glutamate excitation are caused by diverse events, such as mechanical stress, formation of apoptotic and necrotic cells, failure of Na+/K+ ATPase in the axonal membrane, lipid peroxidation and formation of 4-hydroxynonenal[5]. Hyper-activation of NMDA and AMPA receptors increases the influx of Ca2+ and Na+ ions which further promotes apoptosis and necrotic cell death[12].
High levels of glutamate in necrotic cells alter the ionic flux by increasing intracellular Na+ and Ca2+ concentrations and decreasing intracellular K+ concentrations. An increase in Ca2+ concentration inhibits mitochondrial respiration and energy depletion and consequently disturbs ionic homeostasis. Alteration in the function of Na+/K+ ATPase elevates axonal membrane depolarisation and leads to excessive Na+ influx within axon membranes. This ionic dysregulation causes cell cytotoxic oedema, axonal acidosis, increased Ca2+ membrane permeability, activation of phospholipases, increased reactive oxygen species (ROS) generation and mitochondrial dysfunction[11][12](Figure 1c).
Mitochondria are an integral component for cellular metabolism because they generate ATP (Adenosine triphosphate) molecules through phosphorylation. These organelles have four components, i.e. an outer mitochondrial membrane (OMM), inner mitochondrial membrane (IMM), intermembrane space (IMS) and inner matrix. OMM regulates the passage of molecules via voltage-dependent anion channels (VDAC) and maintains a potential of 5 kDa, and IMM controls the exchange of oxygen, water and carbon dioxide[13]. The electron transport chain (ETC) regulates the proton gradient within mitochondria and comprises NADH dehydrogenase (complex 1) and ATP synthases (complex V). Complex 1 oxidises NADH and produces energy. CNS cells contain a large number of complex 1 and generate ROS. Coenzyme Q and cytochrome regulate electron transport in ETC. This transportation and the control of electrons reduce the production of ROS. Complex V generates ATP, acts as a proton channel, converts ADP to ATP and utilises ATP to pump back protons to intermembrane space, hence utilising energy in place of producing ATP[13]. Mitochondria also work as energy reservoirs, regulate cytosolic Ca2+ levels and serve as a vital role in calcium-dependent neuronal death [8]. In SCI, elevated cytosolic Ca2+ levels activate the complex 1, increase ATP generation and promote ROS production. Ca2+ passes the mitochondria through the mitochondrial calcium uniporter[9]. The accumulation of high cytosolic Ca2+ leads to membrane permeabilisation and increases mitochondrial permeability transition pores (mPTPs)[13]. The opening of mPTPs disturbs the proton gradient, inactivates ATP production increases the influx of water and other components within a mitochondrial matrix, and results in cell swelling and finally death[13][14] (Figure 2). Calcium overload also promotes protein kinases and phospholipases which cause calpain-associated protein degradation and oxidative damage[9][15]. Most of the energy required by brain is provided by mitochondria, and sufficient energy is required for neuronal survival; therefore, mitochondrial dysfunction could result in neuronal death[16].
High ROS and reactive nitrogen species (RNS) generation induces various deleterious effects, including lipid peroxidation on different body organs. Lipid peroxidation transpires in three steps: (i) ROS reacts with the membrane’s polyunsaturated fatty acid component and snatches an electron from it. This electron binds to lipid molecules and generates reactive lipid species (ii) which quenches other radicals, generates additional reactive species and (iii) finally produce other reactive species including 4-hydroxynonenal (HNE) and 2-propenal[17]. Neuroinflammation is a key process associated with SCI and involved numerous cell types such as neutrophils, microglia, macrophages, astrocytes, dendritic cells (DCs) and B-and T-lymphocytes and molecular components such as cytokines and prostanoids[17][18]. The complex inflammatory responses following SCI produce neurotoxic or neuroprotective effects depending on the duration and time of responses. Early inflammatory cells and mediators such as macrophages may also have beneficial functions by assisting in inflammation, repair and recovery[18]. Apoptosis and necrosis are vital cell death processes in SCI. In 2012, the Nomenclature Committee on Cell Death lists 12 different types of cell death mechanisms such as necroptosis, pyroptosis, autophagy and netosis[19][20]. During apoptosis, the cell shrinks, followed by phagocytosis[21][22] (Figure 2). Another major process that mediates cell death is autophagy [23] which works as a recycling agent and detoxifies unwanted proteins and organelles by promoting autophagosomal and lysosomal pathways. During SCI, the abnormal activation of autophagosomes and lysosomes triggers rapid cell death[24]. Few other mechanisms of cell death such as programmed cell death called necroptis[25], regulated cell death calledparthanatos[26] and caspase-independent cell death pathways often involving apoptosis-inducing factor (AIF)[21] are not clearly understood and need further investigations. Necroptosis is a programmed necrotic cell death playing a vital role in neuronal cell death[25]. The detailed explanation of ROS and RNS generation, apoptotic pathways and neuroinflammation is presented in the following section.
Acute axonal degeneration (AAD, Figure 3) is another important clinical manifestation of early acute SCI phase. This process induces other effectors such as cysteine protease calpain and Wallerian degeneration which further promote axonal degeneration[27]. AAD is initiated by a high Ca2+ influx into axons. A high Ca2+ deposition increases AAD risk in axons[27]. This phenomenon occurs in two phases, the earlier phase occurs within 15 min post-injury, and the later phase called Wallerian degeneration occurs after a few hours (24–48 h)[28]. The Wallerian degeneration is manifested by the formation of retraction bulbs, a microtubule network that inhibits axonal regeneration[28]. The anterograde degenerative mechanism is termed as Wallerian degeneration; however, retrograde degeneration of axons is termed as axonal dieback[6].
Demyelination occurs when myelin, the protective coating of nerve cells, is damaged. This process slows down the messages sent along axons and deteriorates axon and oligodendrocytes[29]. Oligodendrocytes are myelinating cells that promote the proliferation and myelination of axons[30] and are sensitive to glutamate excitotoxicity that occurs due to the hyperactivation of AMPA, kainate and NMDA receptors [11]. During SCI, oligodendrocytes undergo necrosis and apoptosis. A high glutamate level increases Ca2+ influx that provokes cell death [11]. Damage to oligodendrocytes is also induced by ROS and RNS production, glutathione reduction, and increase in iron load and peroxisome hyperactivation [31]. ROS production by neutrophils and microglia triggers the release of pro-inflammatory cytokines such as TNFα, IL-2 and interferon (IFN) γ and proteases and further facilitates oligodendrocyte apoptosis [31]. The formation of pro-inflammatory cytokines such as TNFα plays a vital role in the inflammation and apoptosis of oligodendrocytes[31]. The apoptosis of oligodendrocytes causes the demyelination of axon and results in the loss of axonal function and stability because single oligodendrocytes myelinate several other axons[31][32]. The demyelination of oligodendrocytes also induces the expression of Fas-receptors that release caspases 3 and 8 which mediate the apoptosis[22][23].
Glial scar formation (gliosis) (Figure 2) is a reactive cellular mechanism that is facilitated by astrocytes and occurs during the chronic secondary phase of SCI. The scarring of astrocytes (astrogliosis) is the body’s natural process that shields and starts the healing post-SCI[33]. Astrocytes are an important component of the nervous system. The astrocytes are sensitive towards changes such as alteration in gene expression, hypertrophy, and excitations[34]. The other major constituents of the scar tissue are pericytes and the connective tissues. In normal physiology, the number of astrocytes is 10 times higher in spinal cord parenchyma that that of pericytes. However, 2 weeks after post-injury, the pericytes are twice the number of astrocytes[34]. Pericytes secrete specific markers that promote fibroblast to express ECM such as fibronectin which serves as the main component of scar connective tissues [35].
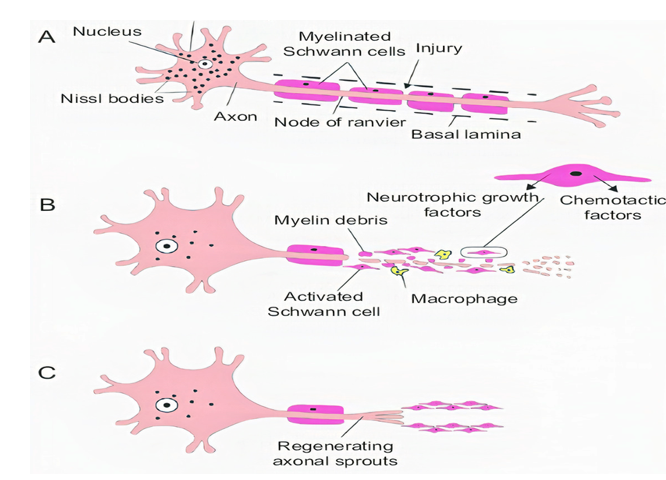
Figure 3. Stages of axon degeneration, (A) acute injury responses, (B) acute axonal degeneration (AAD) and (C) Wallerian degeneration.
The continuous enlargement of lesion site and formation of the cyst is the hallmark feature of SCI. The formation of cyst reveals ongoing apoptotic responses while astrocytes undergo necroptosis cell death through TLR4/MyD88 signalling[36]. Cyst formation leads to syringomyelia in approximately one-third of patients with SCI. Syringomyelia is a condition in which a cyst (syrinx) or cavity develops within the spinal cord, progresses over time and damages the spinal cord. The destruction may result in sensation loss, paralysis, weakness and stiffness in the back, shoulders and extremities[37]. The complications related to syringomyelia are often observed in SCI, but the pathophysiology of syrinx formation is poorly understood[37].
References
- Khorasanizadeh, M.; Yousefifard, M.; Eskian, M.; Lu, Y.; Chalangari, M.; Harrop, J.S.; Rahimi-Movaghar, V. Neurological recovery following traumatic spinal cord injury: A systematic review and meta-analysis. J. Neurosurg. 2019, 30, 683–699.
- Katoh, H.; Yokota, K.; Fehlings, M.G. Regeneration of spinal cord connectivity through stem cell transplantation and biomaterial scaffolds. Front. Cell. Neurosci. 2019, 13, 248.
- Htwe, O.; Hussain, R.I.; Naicker. A.S. Challenges in Managing Severe Lower Limb Spasticity Associated with Bilateral Hip Joints Subluxation. Eur. J. Gen. Med. 2016, 13, 165–167.
- Ohnmar, H.; Das, S.; Naicker, A.S. An interesting case of Autonomic Dysreflexia. Clin. Ter. 2009, 160, 371.
- O’Shea, T.M.; Burda, J.E.; Sofroniew, M.V. Cell biology of spinal cord injury and repair. J. Clin. Investig. 2017, 127, 3259–3270.
- Couillard-Despres, S.; Bieler, L.; Vogl, M. Pathophysiology of traumatic spinal cord injury. Neurological Aspects of Spinal Cord Injury; Springer: Cham, Switzerland, 2017; pp. 503–528.
- Dimitrijevic, M.R.; Danner, S.M.; Mayr, W. Neurocontrol of movement in humans with spinal cord injury. Artif. Organs 2017, 39, 823–833.
- Turtle, J.D.; Henwood, M.K.; Strain, M.M.; Huang, Y.J.; Miranda, R.C.; & Grau, J.W. Engaging pain fibers after a spinal cord injury fosters hemorrhage and expands the area of secondary injury. Experimental neurology, 2019, 311, 115–124.
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front. Neurol. 2019, 10, 282.
- Tran, A.P.; Warren, P.M.; Silver, J. The biology of regeneration failure and success after spinal cord injury. Physiol. Rev. 2018, 98, 881–917.
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246.
- Vanzulli, I.; Butt, A.M. mGluR5 protect astrocytes from ischemic damage in postnatal CNS white matter. Cell Calcium 2015, 58, 423–430.
- Cao, Y.; Lv, G.; Wang, Y.S.; Fan, Z.K.; Bi, Y.L.; Zhao, L.; Guo, Z.P. Mitochondrial fusion and fission after spinal sacord injury in rats. Brain Res. 2013, 1522, 59–66.
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636.
- Hall, E.D. Antioxidant therapies for acute spinal cord injury. Neurotherapeutics 2011, 8, 152–167.
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22.
- Hall, E.D.; Wang, J.A.; Bosken, J.M.; Singh, I.N. Lipid peroxidation in brain or spinal cord mitochondria after injury. J. Bioenerg. Biomembr. 2016, 48, 169–174.
- Miron, V.E.; Franklin, R.J. Macrophages and CNS remyelination. J. Neurochem. 2014, 130, 165–171.
- Jones, T.B. Lymphocytes and autoimmunity after spinal cord injury. Exp. Neurol. 2014, 258, 78–90.
- Galluzzi, L.; Vitale, I.; Abrams; J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120.
- Dunai, Z.; Bauer, P.I.; Mihalik, R. Necroptosis: Biochemical, physiological and pathological aspects. Pathol. Oncol. Res. 2011, 17, 791–800.
- Liu, S.; Li, Y.; Choi, H.M.C.; Sarkar, C.; Koh, E.Y.; Wu, J.; Lipinski, M.M. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018, 9, 476.
- Sobrido-Cameán, D.; Barreiro-Iglesias, A. Role of caspase-8 and Fas in cell death after spinal cord injury. Front. Mol. Neurosci. 2018, 11, 101.
- Yu, W.R.; Fehlings, M.G. Fas/FasL-mediated apoptosis and inflammation are key features of acute human spinal cord injury: Implications for translational, clinical application. Acta Neuropathol. 2011, 122, 747–761, doi:10.1007/s00401-011-0882-3.
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid. Redox Signal. 2015, 23, 565–577.
- Liu, M.; Wu, W.; Li, H.; Li, S.; Huang, L.T.; Yang, Y.Q.; Sun, Q.; Wang, C.X.; Yu, Z.; Hang, C.H. Necroptosis, a novel type of programmed cell death, contributes to early neural cells damage after spinal cord injury in adult mice. J. Spinal Cord Med. 2015, 38, 745–753, doi:10.1179/2045772314y.00000002243127.
- Ginet, V.; Spiehlmann, A.; Rummel, C.; Rudinskiy, N.; Grishchuk, Y.; Luthi-Carter, R.; Clarke, P.G.; Truttmann, A.C.; Puyal, J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014, 10, 846–860.
- Williams, P.R.; Marincu, B.N.; Sorbara, C.D.; Mahler, C.F.; Schumacher, A.M.; Griesbeck, O.; Kerschensteiner, M.; Misgeld, T. A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat. Commun. 2014, 5, 1–11.
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18.
- Cohen-Adad, J.; El Mendili, M.M.; Lehéricy, S.; Pradat, P.F.; Blancho, S.; Rossignol, S.; Benali, H. Demyelination and degeneration in the injured human spinal cord detected with diffusion and magnetization transfer MRI. Neuroimage 2011, 55, 1024–1033.
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front. Cell Dev. Biol. 2016, 4, 71.
- Almad, A.; Sahinkaya, F.R.; McTigue, D.M. Oligodendrocyte fate after spinal cord injury. Neurotherapeutics 2011, 8, 262–273.
- Mekhail, M.; Almazan, G.; Tabrizian, M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: A review. Prog. Neurobiol. 2012, 96, 322–339.
- Yuan, Y.M.; He, C. The glial scar in spinal cord injury and repair. Neurosci. Bull. 2013, 29, 421–435.
- Billakanti, R.; Karimi-Abdolrezaee, S. Reactive astrogliosis after spinal cord injury-beneficial and detrimental effects. Mol. Neurobiol. 2012, 46, 251–264.
- Göritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisén, J. A pericyte origin of spinal cord scar tissue. Science 2011, 333, 238–242.
- Fan, H.; Zhang, K.; Shan, L.; Kuang, F.; Chen, K.; Zhu, K.; Ma, H.; Ju, G.; Wang, Y.Z. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol. Neurodegener. 2016, 11, 14.



