 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Radovan Hojs | + 2085 word(s) | 2085 | 2020-09-28 09:01:11 | | | |
| 2 | Nora Tang | Meta information modification | 2085 | 2020-11-06 04:40:04 | | | | |
| 3 | Nora Tang | Meta information modification | 2085 | 2020-11-06 04:41:51 | | |
Video Upload Options
Oxidative stress is important in the development and progression of diabetic nephropathy (DN). Many pathways and molecules are involved in the induction of oxidative stress in DN. The identification of biomarkers of oxidative stress contibutes to our understanding of development and progression of DN towards end-stage reanal disease.
1. Introduction
Oxidative stress is not only an important factor in the development of type 1 and type 2 diabetes, but it also has a significant role in the development of diabetic complications, including diabetic nephropathy (DN) [1][2][3][4][5][6][7][8][9][10][11][12]. Oxidative stress is linked with metabolic changes and alterations in renal haemodynamics. Both mechanisms have adverse synergistic effects [10]. Oxidative stress is directly linked to podocyte damage, proteinuria, and tubulointerstitial fibrosis [13]. Additionally, vascular oxidative stress has an important role in CKD progression (Figure 1) [13][14][15][16].
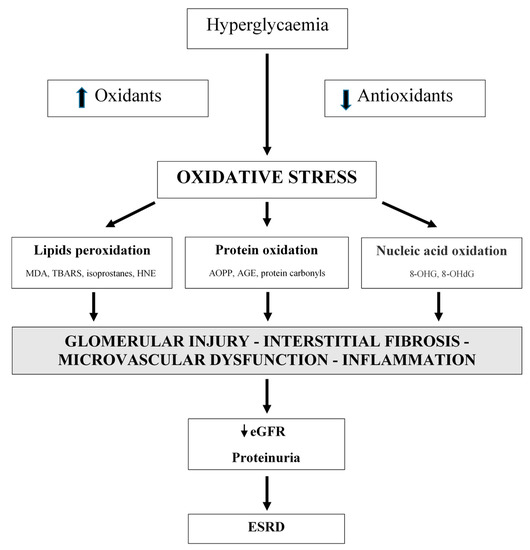
Figure 1. Oxidative stress is a significant factor in the development of diabetic nephropathy. Oxidative stress is associated with metabolic changes and alterations in renal hemodynamic. MDA: malondialdehyde; TBARS: thiobarbituric acid reactive substances; HNE: 4-hydroxynonenal; AOPP: advanced oxidation protein products; AGE: advanced glycation end products; 8-OHG: 8-hydroxyguanosine; 8-OHdG: 8-hydroxy-2′-deoxyguanosine; eGFR: estimated glomerular filtration rate; ESRD: end-stage renal disease.
2. Oxidative Stress and Glomerular Injury
Podocytes are vulnerable to oxidative damage [13]. Mature podocytes are highly differentiated cells and respond to injury with detachment from the glomerular basement membrane, dedifferentiation, autophagy, and apoptosis [17]. An important consequence of podocyte injury is proteinuria, which is a well-known marker of kidney damage and is associated with CKD progression [17][18]. Proteinuria is an important factor in inducing mesangial and tubular toxicity and is involved in local and systemic inflammatory pathways [18][19].
In early studies, it was shown that puromycin aminonucleoside, a podocyte toxin, induced glomerular injury in rats through ROS [13][20][21]. In these studies, antioxidants also provided protection against the changes in podocytes [21]. Later, ROS-mediated DNA damage was also shown [22]. Podocyte injury and dysfunctional glomerular filtration barrier is important in the process of focal segmental glomerular sclerosis (FSGS). The development and progression of FSGS is associated with transforming growth factor beta (TGF-β) activation in podocytes [23]. TGF-β is involved in crosstalk between podocytes and the glomerular endothelium [24]. TGF-β promotes synthesis of endothelin precursors in podocytes and expression of endothelin receptors. The binding of endothelin with its receptors suppresses mitochondrial function and induces oxidative stress in the glomerular endothelium [24]. Mitochondrial oxidative DNA damage was evident before podocyte injury [24].
Other oxidative stress markers are advanced oxidation protein products (AOPPs). They are dityrosine-containing products of plasma proteins [13]. Higher AOPP levels were found in patients with CKD compared to controls [25][26]. Podocyte injury, proteinuria, and glomerulosclerosis were associated with AOPPs through a NOX-dependent mechanism [27]. In normal rats, chronic administration of AOPPs increased proteinuria and urinary 8-hydroxydeoxyguanosine (8-OHdG) excretion. On the other hand, chronic inhibition of NOX by apocynin prevented podocyte apoptosis and decreased proteinuria in these rats [27]. AOPPs interacted with the receptor of advanced glycation end products (RAGE) on podocytes [28]. Additionally, blocking RAGE by anti-RAGE immunoglobulin G or its silencing by siRNA significantly protected podocytes from AOPP-induced apoptosis and ameliorated proteinuria in AOPP-challenged mice [28]. AOPPs are involved in the activation of Wnt/β-catenin signalling. Wnts are a family of secretory proteins that induce a series of signals which results in the phosphorylation of β-catenin [29]. After activation, β-catenin enters the nucleus and promotes the transcription of Wnt target genes [29]. Wnt/β-catenin signalling is silent in normal adults. AOPPs induce NOX activation via plasma membrane receptor RAGE, which promotes the activation of the nuclear factor kappa B (NF-κB) transcription factor. The NF-κB transcription factor leads to the induction of Wnt ligands, such as Wnt1 and Wnt7a, and the activation of β-catenin [30]. Accumulating evidence suggests that Wnt/β-catenin has an important role in oxidative stress-induced podocyte damage and proteinuria [30]. Recently, it was demonstrated that a blockade of Wnt signalling preserves podocyte integrity and ameliorates proteinuria [30]. According to the mentioned data, targeting Wnt/β-catenin could be a new therapeutic modality for proteinuric CKD [30].
In the middle-aged general population, a marker of oxidative DNA damage, urinary 8-hydroxyguanosine (8-OHG) excretion, was independently associated with incident low-grade albuminuria during almost 6 years of follow-up [31].
Additionally, oxidative stress is also associated with progressive renal failure. Finnish-type congenital nephrotic syndrome (NPHS1) is a rare genetic kidney disease caused by mutations in the NPHS1 gene, which codes for the podocyte protein nephrin [32]. The disease is characterised by heavy proteinuria and hypoproteinaemia from birth [32]. In nephrectomised kidneys from children with NPHS1, interstitial expression of MPO was demonstrated [32]. This enzyme generates hypoclorous acid (HOCl), which causes irreversible tissue damage [32]. The concentration of free GSH in the cortex of the NPHS1 kidneys, which is a major antioxidant, was extremely low as compared to controls [32]. All these findings support the fact that proteinuric kidneys are under heavy oxidative stress.
In proteinuric CKD, tubulointerstitial injury with subsequent progressive loss of renal function is common. During urinary albumin endocytosis in the proximal tubule, protein kinase C-dependent NOX-mediated ROS generation is induced and this is responsible for enhanced NF-κB activity and the induction of NF-κB-dependent pathways of interstitial inflammation [33][34].
Less is known about the role of antioxidants in proteinuric CKD. Enzyme superoxide dismutase (SOD) protects the kidney from superoxide. Downregulation of cytosolic CuZn-SOD (SOD1) and extracellular CuZn-SOD (SOD3), but not mitochondrial Mn-SOD (SOD2), was observed in the kidney of KK/Ta-Akita mice that exhibit progressive DN [35]. In this study, no change in renal SOD expression in DN-resistant C57BL/6-Akita mice was observed [35]. In another study, a murine model of adriamycin-induced nephropathy was used. Levels of SOD3 diminished throughout the course of disease progression [36]. Interestingly, similar to findings in mice, a decrease in SOD3 in human CKD biopsy samples was found [36]. The authors concluded that SOD3 protects against proteinuric renal injury in vivo. It offers protection through the inhibition of NOX upregulation and downregulation of pathologic β-catenin signalling [36].
3. Oxidative Stress and Interstitial Fibrosis
Disregarding the initial injury, renal fibrosis is the common final pathway leading to ESRD, and the degree of fibrosis or fibroblast number are robust pathologic markers of progression [37]. Tubulointerstitial fibrosis includes the deposition of interstitial matrix with inflammatory cells, tubular cell loss, fibroblast accumulation, and rarefaction of the peritubular microvasculature [37]. Renal scarring is a result of complex interactions of molecular pathways, growth factors, cytokines, and cells [38][39][40][41].
Fibroblasts/myofibroblasts are most responsible for interstitial matrix accumulation and subsequent structural changes [42]. Collagen-producing myofibroblasts in the kidney can be derived from resident fibroblasts, pericytes, perivascular adventitial, epithelial, and/or endothelial sources [42]. Regardless of the origin of the cells, TGF-β1 is the main molecule responsible for myofibroblast activation with the expression of α-smooth muscle actin (α-SMA), which gives the myofibroblasts their contractility [42][43][44]. TGF-β1 increases the activity of NOX and expression of NOX2 and NOX4, homologues of the NOX family, indicating that this growth factor induces the production of ROS [44]. NOX2 and NOX4 have an important role in the conversion of fibroblasts to myofibroblasts [42][44]. It was shown that inhibition of NOX4 inhibited TGF-β-induced stimulation of NOX activity and reduced α-SMA expression [44]. Additionally, inhibition of TGF-β receptor type I reduced TGF-β-enhanced NOX activity and decreased expression of NOX4 and α-SMA [44].
As was shown, NOX synthesises ROS that are involved in fibrosis progression. On the other hand, their effect on renal disease progression is not well understood. In the model of chronic renal injury due to unilateral urinary obstruction, leading to renal fibrosis, wild-type and NOX4-deficient mice were used [45]. In the NOX4-deficient mice, more interstitial fibrosis was found in the obstructed kidney compared to the wild-type mice [45]. More TGF-β1-mediated tubular apoptosis, reduced expression of hypoxia-inducible factor-1α, and vascular endothelial growth factor was also found in the obstructed kidneys of the NOX4-deficient mice [45]. It was shown that the absence of NOX4 increases interstitial kidney fibrosis, independent of NOX2. [45]. NOX4 deficiency increased fibrosis due to enhanced tubular cell apoptosis, decreased microvascularisation, and enhanced oxidative stress [45]. The NOX4-mediated protection might be a consequence of Nrf2 pathway upregulation [46]. The Nrf2/Keap1 system controls the expression of antioxidant genes [46]. Furthermore, Nrf2 plays a protective role in CKD animal models, including DN [47][48].
Uraemic toxins are also involved in the progression of CKD. In the last decade, indoxyl sulphate (IS) and p-cresyl sulphate (PCS), which accumulate with CKD progression, have appeared as key nephrotoxins [49][50]. IS and PCS enhance ROS production in renal tubular cells, which activate the NF-kB pathway, resulting in both oxidative stress and inflammation [50][51]. These mechanisms have been confirmed in studies showing that fibrosis of renal tubules and oxidative stress are significantly enhanced after toxin administration and suppressed after IS reduction [50][51][52]. Additionally, it was shown that antioxidant treatment dose-dependently inhibits the fibrotic and oxidative effects of IS and PCS [53][54].
Recently, it was demonstrated that oxidative stress and autophagy are involved in kidney health and disease [55]. Autophagy is a crucial cellular homeostatic process that cells use to degrade and recycle cellular proteins and remove damaged organelles. It involves the formation of double membrane-bound vesicles called autophagosomes, which later fuse with lysosomes [56]. Basal levels of redox signalling and autophagy signalling are necessary to maintain cellular homeostasis. Under distinct circumstances, changes in autophagic flux have been shown to regulate ROS formation and redox signalling [55]. It is also suggested that ROS and RNS induce autophagy and vice versa [55][57].
4. Oxidative Stress and Microvascular Dysfunction
The endothelium is a fundamental layer in the arterial wall and is essential for the regulation and maintenance of normal renal function [13][14]. Oxidative stress is related to endothelial dysfunction and plays a critical role in CKD progression [14][15][58]. The endothelium secretes nitric oxide (NO), which is produced from arginine by the enzyme NOS [58]. NO is involved in several biological processes, including vasodilatation in smooth muscle cells, inflammation, and immune responses [58]. NOS is expressed as various isoforms: endothelial NOS (eNOS), neuronal NOS (nNOS), inducible NOS (iNOS), and constitutive NOS (cNOS); all have been isolated from the kidney [13][58][59]. The cNOS is expressed in the vessels, glomeruli, and tubules, iNOS is expressed in vascular smooth muscle cells and the mesangium, and eNOS is associated with the vascular endothelium [15][58][60]. Low levels of NO in the endothelium induce the expression of antioxidative genes and protect renal endothelial and mesangial cells from apoptosis and fibrosis but, on the other hand, increased levels of ROS reduce the production of NO via inhibition and/or uncoupling of NOS enzymes [15][58][59][60]. The NO production in the kidney can be blocked by NOS inhibition with asymmetric dimethylarginine (ADMA). ADMA is a natural product formed by the methylation of arginine which accumulates in the plasma of CKD patients in the early stages of CKD [15][59]. The decrease in NO leads to an increase in vascular resistance [59]. Additionally, it was shown in patients with CKD stages 1-5 that levels of serum ADMA and oxidative stress markers (plasma malondialdehyde (MDA), erythrocyte SOD, and GSH-Px) were directly associated with CKD stages [15]. It was shown that the glomerular filtration rate correlated negatively with plasma MDA and ADMA levels and positively with erythrocyte SOD and GSH-Px [15]. Patients with CKD, compared to a control group of healthy subjects, had higher levels of MDA and ADMA and lower levels of erythrocyte SOD and GSH-Px [15]. Furthermore, it was shown that levels of oxidative stress markers and ADMA are independently associated with endothelial function [15].
Autoregulation is important in maintaining renal blood flow, glomerular filtration rate, and tubular fluid flow over a wide range of perfusion pressures. It is dependent on afferent arteriole contraction followed by a tubuloglomerular feedback[61][62]. Impairment of renal autoregulation is associated with CKD progression. In experimental studies, it was documented that ROS mediate myogenic responses of afferent arterioles in CKD models [63]. It was also shown that NOX2 plays an important role in regulating tone and reactivity of afferent arterioles, also in response to angiotensin II (ANG II) and/or adenosine [64]. NOX2-derived ROS scavenges NO, causing subsequent NO deficiency [64]. It was demonstrated that an increase in perfusion pressure increases superoxide (O2•−) in afferent arterioles in normal mice or mice with a genetic deletion of SOD and is involved in the myogenic contractions of afferent arterioles [65][66]. H2O2 impaired autoregulation of afferent arterioles in five out of six nephrectomised mice [62][65][66].
5. Oxidative Stress and Chronic Inflammation
Oxidative stress and inflammation, as well as their interaction, have an important role in the pathogenesis and progression of CKD [67]. Both promote renal injury through damage of molecular components [68]. The primary pathological mechanism that links oxidative stress, inflammation, and CKD progression includes an initial injury to the kidney by intra- and extracellular oxygen-derived radicals and the resultant inflammation [68]. In recent years, some important review papers have been published showing the importance of inflammation in the pathogenesis and progression of CKD [3][7][69][13][70][71][72][73][74].
References
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature. Hypertension 2003, 42, 1075–1081.
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673.
- Elmarakby, A.A.; Sullivan, J.C. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther. 2012, 30, 49–59.
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 2014, 1840, 2709–2729.
- Brown, W.V. Microvascular complications of diabetes mellitus: Renal protection accompanies cardiovascular protection. Am. J. Cardiol. 2008, 102, 10L–13L.
- Wu, J.; Mei, C.; Vlassara, H.; Striker, G.E.; Zheng, F. Oxidative stress-induced JNK activation contributes to proinflammatory phenotype of aging diabetic mesangial cells. Am. J. Physiol. Renal Physiol. 2009, 297, F1622–F1631.
- García-García, P.M.; Getino-Melián, M.A.; Domínguez-Pimentel, V.; Navarro-González, J.F. Inflammation in diabetic kidney disease. World J. Diabetes 2014, 5, 431–443.
- Wolf, G. New insights into the pathophysiology of diabetic nephropathy: From hemodynamics to molecular pathology. Eur. J. Clin. Investig. 2004, 34, 785–796.
- Kanwar, Y.S.; Wada, J.; Sun, L.; Xie, P.; Wallner, E.I.; Chen, S.; Chugh, S.; Danesh, F.R. Diabetic nephropathy: Mechanisms of renal disease progression. Exp. Biol. Med. 2008, 233, 4–11.
- Sifuentes-Franco, S.; Padilla-Tejeda, D.E.; Carrillo-Ibarra, S.; Miranda-Díaz, A.G. Oxidative stress, apoptosis, and mitochondrial function in diabetic nephropathy. Int. J. Endocrinol. 2018, 2018, 1875870.
- Ceriello, A.; Morocutti, A.; Mercuri, F.; Quagliaro, L.; Moro, M.; Damante, G.; Viberti, G.G. Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes 2000, 49, 2170–2177.
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9.
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711.
- Zoccali, C. The endothelium as a target in renal diseases. J. Nephrol. 2007, 20, S39–S44.
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50.
- Kielstein, J.T.; Böger, R.H.; Bode-Böger, S.M.; Frölich, J.C.; Haller, H.; Ritz, E.; Fliser, D. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J. Am. Soc. Nephrol. 2002, 13, 170–176.
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230.
- Coresh, J.; Heerspink, H.J.L.; Sang, Y.; Matsushita, K.; Arnlov, J.; Astor, B.C.; Black, C.; Brunskill, N.J.; Carrero, J.J.; Feldman, H.I.; et al. Change in albuminuria and subsequent risk of end-stage kidney disease: An individual participant-level consortium meta-analysis of observational studies. Lancet Diabetes Endocrinol. 2019, 7, 115–127.
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150.
- Gwinner, W.; Landmesser, U.; Brandes, R.P.; Kubat, B.; Plasger, J.; Eberhard, O.; Koch, K.M.; Olbricht, C.J. Reactive oxygen species and antioxidant defense in puromycin aminonucleoside glomerulopathy. J. Am. Soc. Nephrol. 1997, 8, 1722–1731.
- Ricardo, S.D.; Bertram, J.F.; Ryan, G.B. Antioxidants protect podocyte foot processes in puromycin aminonucleoside-treated rats. J. Am. Soc. Nephrol. 1994, 4, 1974–1986.
- Marshall, C.B.; Pippin, J.W.; Krofft, R.D.; Shankland, S.J. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int. 2006, 70, 1962–1973.
- Bottinger, E.P. TGF-β in renal injury and disease. Semin. Nephrol. 2007, 27, 309–320.
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621.
- Witko-Sarsat, V.; Friedlander, M.; Capeillère-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stressin uremia. Kidney Int. 1996, 49, 1304–1313.
- Witko-Sarsat, V.; Gausson, V.; Descamps-Latscha, B. Are advanced oxidation protein products potential uremic toxins? Kidney Int. Suppl. 2003, 84, S11–S14.
- Zhou, L.L.; Hou, F.F.; Wang, G.B.; Yang, F.; Xie, D.; Wang, Y.P.; Tian, J.W. Accumulation of advanced oxidation protein products induces podocyte apoptosis and depletion through NADPH dependent mechanisms. Kidney Int. 2009, 76, 1148–1160.
- Zhou, L.L.; Cao, W.; Xie, C.; Tian, J.; Zhou, Z.; Zhou, Q.; Zhu, P.; Li, A.; Liu, Y.; Miyata, T.; et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int. 2012, 82, 759–770.
- Zhou, L.; Liu, Y. Wnt/β-catenin signaling and podocyte dysfunction in proteinuric kidney disease. Nat. Rev. Nephrol. 2015, 11, 535–545.
- Zhou, L.; Chen, X.; Lu, M.; Wu, Q.; Yuan, Q.; Hu, C.; Miao, J.; Zhang, Y.; Li, H.; Hou, F.F.; et al. Wnt/β-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int. 2019, 95, 830–845.
- Schei, J.; Fuskevåg, O.M.; Stefansson, V.T.N.; Solbu, M.D.; Jenssen, T.G.; Eriksen, B.O.; Melsom, T. Urinary markers of oxidative stress Are Associated with Albuminuria but Not GFR Decline. Kidney Int. Rep. 2017, 3, 573–582.
- Kuusniemi, A.M.; Lapatto, R.; Holmberg, C.; Karikoski, R.; Rapola, J.; Jalanko, H. Kidneys with heavy proteinuria show fibrosis, inflammation, and oxidative stress, but no tubular phenotypic change. Kidney Int. 2005, 68, 121–132.
- Morigi, M.; Macconi, D.; Zoja, C.; Donadelli, R.; Buelli, S.; Zanchi, C.; Ghilardi, M.; Remuzzi, G. Protein overload-induced NF-kappaB activation in proximal tubular cells requires H2O2 through a PKC-dependent pathway. J. Am. Soc. Nephrol. 2002, 13, 1179–1189.
- Souma, T.; Abe, M.; Moriguchi, T.; Takai, J.; Yanagisawa-Miyazawa, N.; Shibata, E.; Akiyama, Y.; Toyohara, T.; Suzuki, T.; Tanemoto, M.; et al. Luminl alkalinization attenuates proteinuria induced oxidative damage in proximal tubular cells. J. Am. Soc. Nephrol. 2011, 22, 635–648.
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 1303–1313.
- Tan, R.J.; Zhou, D.; Xiao, L.; Zhou, L.; Li, Y.; Bastacky, S.I.; Oury, T.D.; Liu, Y. Extracellular superoxide dismutase protects against proteinuric kidney disease. J. Am. Soc. Nephrol. 2015, 26, 2447–2459.
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834.
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36.
- Tampe, B.; Zeisberg, M. Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol. Dial. Transplant. 2014, 29, iv72–iv79.
- Lv, W.; Booz, G.W.; Fan, F.; Wang, Y.; Roman, R.J. Oxidative stress and renal fibrosis: Recent insights for the development of novel therapeutic strategies. Front. Physiol. 2018, 16, 105.
- Okamura, D.M.; Pennathur, S. The balance of powers: Redox regulation of fibrogenic pathways in kidney injury. Redox Biol. 2015, 6, 495–504.
- Barnes, J.L.; Gorin, Y. Myofibroblast diferentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956.
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307.
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1 induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102.
- Nlandu Khodo, S.; Dizin, E.; Sossauer, G.; Szanto, I.; Martin, P.Y.; Feraille, E.; Krause, K.H.; de Seigneux, S. NADPH oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol. 2012, 23, 1967–1976.
- Zoja, C.; Benigni, A.; Remuzzi, G. The Nrf2 pathway in the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i19–i24.
- Choi, B.H.; Kang, K.S.; Kwak, M.K. Effect of redox modulating NRF2 activators on chronic kidney disease. Molecules 2014, 19, 12727–12759.
- Soetikno, V.; Sari, F.R.; Lakshmanan, A.P.; Arumugam, S.; Harima, M.; Suzuki, K. Curcumin alleviates oxidative stress, inflammation, and renal fibrosis in remnant kidney through the Nrf2-keap1 pathway. Mol. Nutr. Food Res. 2013, 57, 1649–1659.
- Meijers, B.K.I.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761.
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3–4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317.
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592.
- Lekawanvijit, S.; Kompa, A.R.; Manabe, M.; Wang, B.H.; Langham, R.G.; Nishijima, F.; Kelly, D.J.; Krum, H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS ONE 2012, 7, e41281.
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-kappaB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680.
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39.
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214.
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell. Biol. 2007, 9, 1102–1109.
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427.
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004, 24, 354–365.
- Aldámiz-Echevarría, L.; Andrade, F. Asymmetric dimethylarginine, endothelial dysfunction and renal disease. Int. J. Mol. Sci. 2012, 13, 11288–11311.
- Sullivan, J.C.; Pollock, J.S. Coupled and uncoupled NOS: Separate but equal? Uncoupled NOS in endothelial cells is a critical pathway for intracellular signaling. Circ. Res. 2006, 98, 717–719.
- Just, A. Mechanisms of renal blood flow autoregulation: Dynamics and contributions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1–R17.
- Lai, E.Y.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. Superoxide modulates myogenic contractions of mouse afferent arterioles. Hypertension 2011, 58, 650–656.
- Li, L.; Lai, E.Y.; Luo, Z.; Solis, G.; Griendling, K.K.; Taylor, W.R.; Jose, P.A.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. Superoxide and hydrogen peroxide counterregulate myogenic contractions in renal afferent arterioles from a mouse model of chronic kidney disease. Kidney Int. 2017, 92, 625–633.
- Carlström, M.; Lai, E.Y.; Ma, Z.; Patzak, A.; Brown, R.D.; Persson, A.E. Role of NOX2 in the regulation of afferent arteriole responsiveness. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R72–R79.
- Li, L.; Lai, E.Y.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. Differential effects of superoxide and hydrogen peroxide on myogenic signaling, membrane potential, and contractions of mouse renal afferent arterioles. Am. J. Physiol. Renal. Physiol. 2016, 310, F1197–F1205.
- Lai, E.Y.; Solis, G.; Luo, Z.; Carlstrom, M.; Sandberg, K.; Holland, S.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. p47phox is required for afferent arteriolar contractile responses to angiotensin II and perfusion pressure in mice. Hypertension 2012, 59, 415–420.
- Xu, G.; Luo, K.; Liu, H.; Huang, T.; Fang, X.; Tu, W. The progress of inflammation and oxidative stress in patients with chronic kidney disease. Ren. Fail. 2015, 37, 45–49.
- Tucker, P.S.; Scanlan, A.T.; Dalbo, V.J. Chronic kidney disease influences multiple systems: Describing the relationship between oxidative stress, inflammation, kidney damage, and concomitant disease. Oxid. Med. Cell. Longev. 2015, 2015, 806358.
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991.
- Rivero, A.; Mora, C.; Muros, M.; García, J.; Herrera, H.; Navarro-González, J.F. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin. Sci. 2009, 116, 479–492.
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Belén Sanz, A.; Herencia, C.; Mezzano, S.; et al. Pathogenic pathways and therapeutic approaches targeting inflammation in diabetic nephropathy. Int. J. Mol. Sci. 2020, 21, 3798.
- Pérez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in Diabetic Kidney Disease. Nephron 2019, 143, 12–16.
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J. Immunol. Res. 2018, 2018, 2180373.
- Qian, Q. Inflammation: A key contributor to the genesis and progression of chronic kidney disease. Contrib. Nephrol. 2017, 191, 72–83.


