 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Negroni | + 7557 word(s) | 7557 | 2020-10-13 07:50:40 | | | |
| 2 | Bruce Ren | -3936 word(s) | 3621 | 2020-10-20 04:46:35 | | | | |
| 3 | Bruce Ren | Meta information modification | 3621 | 2020-10-20 10:54:21 | | |
Video Upload Options
Necroptosis is a caspases-independent programmed cell death displaying intermediate features between necrosis and apoptosis. Albeit some physiological roles during embryonic development such tissue homeostasis and innate immune response are documented, necroptosis is mainly considered a pro-inflammatory cell death. Key actors of necroptosis are the receptor-interacting-protein-kinases, RIPK1 and RIPK3, and their target, the mixed-lineage-kinase-domain-like protein, MLKL. The intestinal epithelium has one of the highest rates of cellular turnover in a process that is tightly regulated. Altered necroptosis at the intestinal epithelium leads to uncontrolled microbial translocation and deleterious inflammation. Indeed, necroptosis plays a role in many disease conditions and inhibiting necroptosis is currently considered a promising therapeutic strategy.
1. Introduction
Cell death is crucial during the development and maintenance of tissue homeostasis in multicellular organisms. Until recently, apoptosis was considered the only molecularly controlled type of cell death, as opposed to necrosis, which was classically described as nonregulated accidental cell death. Several types of programmed necrotic cell death have been recently identified, which, although sharing common morphological features, such as cellular volume increase, organelles swelling, and plasma membrane disruption, however, respond to different triggers and follow distinct biochemical pathways [1]. Necroptosis is a programmed necrosis executed by the activation of death receptors including tumor necrosis factor receptor 1 (TNFR1), Fas receptor (FasR), TNF-related apoptosis-inducing ligand–death receptor 1 (TRAILR1), interferon receptor (IFNR) and also pattern-recognition receptors (PRRs) such as Toll-like receptors (TLRs) and RIG-like receptors (RLRs) [2][3][4].
The receptor-interacting protein kinase 1 (RIPK1) and 3 (RIPK3), as well as their target, the mixed lineage kinase domain-like protein (MLKL), are required to initiate necroptosis [5][6][7]. Necroptosis is associated with various human diseases including ischemic reperfusion injury, inflammatory, neurodegenerative, infectious, autoimmune diseases and cancer [8][9][10][11][12][13][14][15][16][17][18]. Recently, necroptosis has been also involved in mediating organ rejection in cardiac and renal allografts [19][20][21][22].
Despite the pathological connotation that characterized necroptosis at the beginning of its identification, emerging evidence points to its crucial role in physiological phenomena such as development, immunology and differentiation. For example, mutant mice with kinase-dead RIPK1 or RIPK3 and MLKL deficiency show no detrimental phenotype with regard to development and adult homeostasis. Furthermore, several authors report that necroptosis is able to induce the innate immune response to viral infections, when the virus inhibits host apoptotic machinery and cells are committed to alternative cell death to limit viral replication or generate an immune response [23][24][25][26][27]. Necroptosis is also implicated in the regulation of antigen-activated T-cell proliferation and survival [28][29].
2. Necroptosis Signaling Pathway
Necroptosis is a cellular response to environmental stress that can be caused by chemical and mechanical injury, inflammation, or infection. In addition to traditional triggers (Figure 1), recent evidence shows that necroptosis can be also induced by DNA damage, environmental stresses, such as the hypoxia and glucose level, and chemotherapeutic agents [30][31][32][33][34].
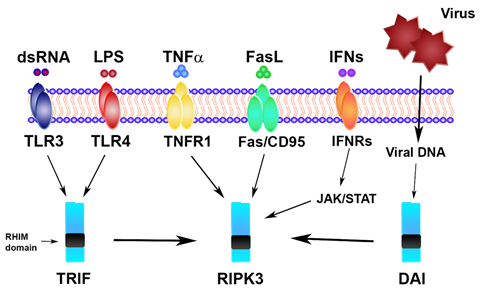
Figure 1. Necroptosis signaling triggers. TLR, Toll-like receptor; LPS, lipopolysaccharide; TNFα, tumor necrosis factor alpha; TNFR1, tumor necrosis factor receptor; FasL, Fas ligand; IFN, interferon; IFNR, interferon receptor; TRIF, Toll/interleukin-1 receptor (TIR) domain-containing interferon-β; DAI, DNA-dependent activator of IFN regulatory factors; RHIM, RIP-homotypic-interaction-motif domain; RIPK3, receptor-interacting protein kinase 3.
The mechanisms underlying the necroptosis pathway are mostly elucidated by a model of tumor necrosis factor alpha (TNFα)-induced cell death [35][36][37][38]. The binding of TNF to TNFR1 induces a conformational change in TNFR1 trimers, leading to the recruitment of a multiprotein platform named complex I, which includes RIPK1, TRADD (TNFR-associated death domain), cIAP1 (cellular inhibitor of apoptosis protein 1), cIAP2 and TRAF (TNFR-associated factor)2. The polyubiquitination of RIPK1 by cIAP1/2 causes the recruitment of complexes IKK (IKKα, IKKβ and NEMO) and TAK1 (TAK1, TAB1 and TAB2), leading to the activation of the transcription factor NF-kB and cell survival. The deubiquitination of RIPK1 by deubiquitinase cylindromatosis (CYLD) or the A20 ubiquitin-editing complex marks the transition from complex I to complex II and inhibits NF-κB activation [39]. TRADD and RIPK1 dissociate from TNFR1 recruiting Fad- associated protein with death domain (FADD) and pro-caspase-8 and leading to the formation of complex IIa, which results in the activation of caspase-8 cleavage followed by apoptosis [40].
The enrollment of RIPK3 causes the formation of complex IIb. Recent scientific advances have revealed the role of the cellular FLICE-inhibitory protein (cFLIP) as a critical switch to control cell survival and death in various tissues. cFLIP protein is evolutionarily conserved and expressed as functionally different isoforms, named long FLIP(L) and short FLIP(S) splice forms. A heteromeric complex between FLIP(L) and caspase-8 prevents the interaction of RIPK1 and RIPK3 in complex IIb, thereby preventing the induction of RIPK3-dependent necroptosis. In contrast, the binding of cFLIP(S) to FADD prevents the recruitment of caspase-8 and apoptosis [41][42][43]. Thus, the activity of caspase-8 is essential in determining the fate of a cell. A defect in the FADD-caspase-8 signaling pathway sensitizes cells towards necroptosis by promoting the assembly of the necrosome while preventing apoptosis. High levels of RIPK3 or caspase-8 inactivation, due to pharmaceutical or genetic intervention, promote the formation of the necrosome which is stabilized by the interaction and trans/autophosphorylation between RIPK1 and RIPK3. The RIP homotypic interaction motif (RHIM) domains on RIPK3 and RIPK1 are essential for their interaction. Interestingly, other RHIM-containing proteins, such as Toll/interleukin-1 receptor (TIR)-domain-containing adapter-inducing interferon-ß (TRIF) and the DNA-dependent activator of interferon-regulatory factor (DAI, also known as ZBP1), may interact and activate RIPK3, regardless of RIPK1, leading to necroptosis [35]. Afterwards, RIPK3 phosphorylates its substrate MLKL that oligomerizes and translocates to the plasma membrane to execute necroptosis [44][45][46][47][48].
Recently, other effectors of necroptosis, downstream RIPK3, have been identified in addition to MLKL, such as the mitochondrial phosphoglicerate mutase 5 (PGAM5), [49] and Ca2+/calmodulin-dependent protein kinase (CAMK)-II [50]. PGAM5 exists as two splicing variants, the long form (PGAM5L) and the short form (PGAM5S). RIPK3, once activated, phosphorylates PGAM5L and then engages PGAM5S on the mitochondrial membrane. Activated PGAM5L/PGAM5S promote mitochondrial fission through the dephosphorylation of dynamin-related protein-1 (DRP1), thus leading to cellular necroptosis. CAMK-II is also phosphorylated by RIPK3 and once activated, regulates multiples ion channels opening with a consequent influx of extracellular ions and eventual plasma membrane damage, independently of MLKL [50] (Figure 2).
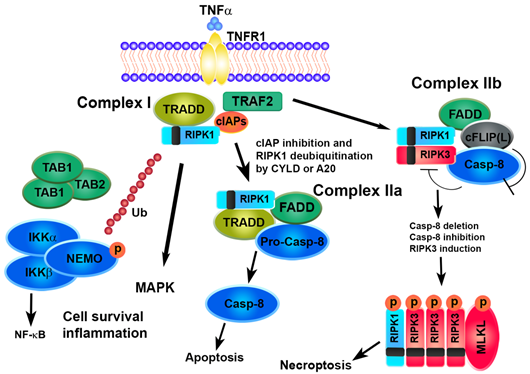
Figure 2. Survival or cell death pathways activated by TNFα/TNFR1. TNFα, tumor necrosis factor alpha; TNFR1, tumor necrosis factor receptor; TRADD, TNFR-associated death domain; TRAF2, TNFR-associated factor, cIAPs, cellular inhibitors of apoptosis protein, RIPK, receptor-interacting protein kinase, TAK1, transforming growth factor-activated kinase 1; TAB, TAK1-binding protein; IKK, inhibitor of NF-kB kinase; NEMO, NF-kB essential modulator; FADD, Fas-associated protein with death domain; CYLD, cylindromatosis lysine 63 deubiquitinase; cFLIP, cellular FLICE-like inhibitory protein; MLKL, mixed lineage kinase domain-like protein.
Necroptosis is finely controlled by several negative regulators in cells. For example, TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation [51][52]. Moreover, absent, transient and sustained levels of TAK1-mediated RIPK1 phosphorylation may represent distinct states in the TNFR1 signaling complex to dictate the activation of alternative cell death mechanisms [53]. The carboxyl terminus of Hsp70-interacting protein (CHIP, also known as STUB1) is a bona fide negative regulator of the RIPK1–RIPK3 necrosome formation leading to the desensitization of TNF-mediated necroptosis [54]. A20 (also known as TNFAIP3, TNF-induced protein 3) and ABIN-1 (also known as TNIP1, TNFAIP3-interacting protein 1), candidate susceptibility genes for several autoimmune or inflammatory diseases, synergistically restrict death by inhibiting TNF-induced caspase-8 activation and RIPK1 kinase activity by blocking both apoptosis and necroptosis [55].
Mice deficient in SET-domain-bifurcate-1 (SETDB1), a histone methyltransferase that mediates the trimethylation of histone H3 at lysine 9, develop necroptosis suggesting a suppressive role for the gene. Accordingly, SETDB1 is downregulated in patients with inflammatory bowel disease (IBD) [56].
3. Interplay between Necroptosis, Apoptosis, Pyroptosis and Autophagy
The interplay between necroptosis and other important cellular processes, such as apoptosis, autophagy or pyroptosis, and the identification of their converging points are crucial for developing novel therapeutic approaches in inflammatory diseases and cancer. Apoptosis occurs during the development as a homeostatic mechanism but also as a defense tool in immune reactions or in the presence of damaged cells [57].
In most situations, apoptosis is the default cell death modality, whereas necroptosis intervenes when key apoptotic mediators are blocked by pharmacological inhibition or genetic ablation or after a cellular stress such as energy ATP depletion or in certain viral infections [58][59].
Caspase-8 represents the molecular switch to control the balance between apoptosis and necroptosis [60][61]. Mice with the intestinal epithelial cell (IEC)-specific deletion of caspase-8 or its adapter FADD develop colitis and ileitis with a loss of Paneth cells and these effects are rescued by RIPK3 deficiency, underlying the importance of necroptosis in driving the pathology in both the small and large intestine [62][63].
Moreover, in mice with IECs, the specific deletion of caspase-8, the stimulation of TLR3 or TLR4 with microbial molecules (poly(I:C) and LPS) or after Salmonella Typhimurium infection causes an RIPK3-dependent epithelial necroptosis with more severe mortality and tissue damage [64][65]. Mice with IEC-specific FADD or caspase-8 deficiency develop colitis depending on MLKL-mediated epithelial cell necroptosis. Besides, caspase-8 and gasdermin-D (GSDMD), the effector of pyroptosis, a highly inflammatory form of regulated cell death which occurs most frequently upon infection with intracellular pathogens, are both required for the development of MLKL-independent ileitis in mice with epithelial FADD deficiency [66].
The execution of pyroptosis initiates the formation of a large supramolecular complex termed the inflammasome, and is regulated via caspases 1 and 11 which results in the activation of GSDMD and following pore formation in the plasma membrane with consequent citoplasmic swelling and the release of intracellular contents, including IL-1β and IL-18 [67].
Recent evidence reports that RIPK3 is able to activate the NLRP3 inflammasome-mediated pyroptosis to drive inflammation [68][69]. Moreover, MLKL protects from Salmonella infection promoting intestinal epithelial barrier function by inducing inflammasome activation in IECs. Besides, mice with MLKL ablation are more susceptible to Salmonella infection and present impaired caspase-1 and GSDMD cleavage with consequently decreased interleukin IL-18 release [70].
Transgenic (Tg) mice, wherein CFLARs, the gene encoding cFLIP, was integrated onto the X chromosome, die perinatally due to severe ileitis. The deletion of RIPK3 or MLKL prevented both necroptosis and apoptosis, and rescued the lethality of the CFLARs Tg mice [71].
A connection between necroptosis and autophagy was also discovered, albeit the molecular mechanisms remain poorly defined [72][73]. Autophagy regulates the degradation of cytoplasmic proteins and organelles within lysosomes, provides nutrients and energy under various stresses, including starvation, cellular and tissue remodeling, and cell death [74]. Recently, it has been shown that autophagy machinery can control programmed cell death, switching between apoptosis and necroptosis, by serving as a scaffold rather than as a degrading cellular material [75].
Moreover, the autophagy gene ATG16L1 is essential in the intestinal epithelium for preventing the loss of Paneth cells and a variant of ATG16L1 is associated with poor survival in allogeneic hematopoietic stem cell transplant recipients and Crohn’s disease (CD), one of the two forms of IBD. Intestinal organoids lacking ATG16L1 reproduce this loss in Paneth cells and display TNFα-mediated necroptosis, indicating that, in contrast to tumor cells in which autophagy promotes caspase-independent cell death, ATG16L1 maintains the intestinal barrier by inhibiting necroptosis in the epithelium [76]. It was also shown that ATG16L1 plays a role in coordinating the outcome of IL-22 signaling in the intestinal epithelium, leading to transient endoplasmic reticulum (ER) stress and the subsequent activation of type I interferon (IFN-I) signaling that amplifies epithelial TNF production and contributes to necroptotic cell death [77]. Besides, a critical function has recently emerged for the mammalian target of rapamycin (mTOR), an evolutionarily conserved protein kinase controlling the balance between cell growth and autophagy, in the regulation of RIPK3 expression and necroptosis in the gut epithelium. Epithelial mTOR hyperactivation, associated with the Western diet, dysbiosis, or the ablation of its repressor TSC1, inhibits RIPK3 degradation in autophagolysosome and promotes the necroptosis of epithelial cells, barrier disruption and chronic inflammation. Accordingly, hyperactive mTOR and aberrant necroptosis were intertwined in humans IBD [78].
Furthermore, the immunity-related GTPase family M protein (IRGM), that plays a role in innate immunity by regulating autophagy in response to several intracellular pathogens, regulates necroptosis and the release of damage-associated molecular patterns (DAMPs) inducing gastrointestinal inflammation. Interestingly, IRGM has been identified in genome-wide association studies as a genetic risk factor in CD. IRGM overexpression by phosphorylated eukaryotic translation initiation factor 2 (pEIF2A) plays a major role as a checkpoint allowing host cell survival by autophagy or host cell elimination by necroptosis [79].
Similarly, necroptosis may promote or repress autophagy, although the mechanisms are still unclear [80]. For example, necroptotic stimulation seems to reduce autophagic activity, as shown by the enlarged puncta of the autophagic substrate Sequestosome 1 (SQSTM1/p62) and its increased colocalization with microtubule-associated protein 1A/1B-light chain 3 (LC3), attenuating autophagic flux before the lysosome fusion step [81]. The activation of necroptosis in mouse dermal fibroblasts and HT-29 human colorectal cancer cells results in the accumulation of the autophagic marker, lipidated LC3B, in an MLKL-dependent manner. Unexpectedly, the necroptosis-induced increase in lipidated LC3B is due to the inhibition of the autophagic flux, not to the activation of autophagy [82].
4. Necroptosis and Inflammation
Molecular mechanisms of cell death have regulatory roles in inflammation and molecular changes associated with different forms of cell death may affect the course of the inflammation in different ways [83]. In recent decades, emerging knowledge on cell death and inflammation has enriched our understanding of the signaling pathways that mediate various programs of cell death and multiple types of inflammatory responses.
Necroptosis is typically considered a highly pro-inflammatory mode of cell death, due to the release of DAMPs, which can promote inflammation and activate an immune response, either alone (in the context of sterile injury) or in combination with pathogen-associated molecular patterns (PAMPs) [84][85]. Necroptosis promotes the activation of macrophages and dendritic cells which increases the levels of pro-inflammatory cytokines, including the IL-1 superfamily, thus triggering acute and chronic inflammatory diseases [84][85].
Recent studies reveal unexpected complexity in the regulation of cell death programs by RIPK1 and RIPK3 with the possibility that necroptosis is one mechanism by which these kinases promote inflammation [86]. For example, RIPK3 seems to control a separate, necrosis-independent pathway of inflammation by regulating NF-κB activation, dendritic cell (DC) biology, innate inflammatory-cytokine expression, and injury-induced tissue repair [87]. Several studies report that RIPK3 mediates the formation of NATCH, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome and following the activation of caspase-1 and -11, through the interaction with different molecules, including caspase-8 and LPS [87][88][89][90]. It is unclear whether MLKL is involved in this process [87]. However, a recent paper showed that MLKL promoted intestinal epithelial barrier function by enhancing inflammasome activation.
In contrast to the traditional view of a pro-inflammatory mode of necroptosis, some authors suggest that it might have anti-inflammatory effects in certain settings, through curbing excessive TNF- or TLR-induced inflammatory cytokine production [91][92][93].
5. Necroptosis and Intestinal Diseases
5.1. Inflammatory Bowel Disease (IBD)
IBD is a chronic, debilitating intestinal disease with various clinical presentations whose main forms are ulcerative colitis (UC) and CD. The cause of IBD is still unknown, although it is believed to be a combination of multiple environmental factors along with genetic inheritance patterns, all leading to an excessive and abnormal immune response against commensal gut flora [94].
The first demonstration of the involvement of necroptosis in the pathogenesis of IBD derives from evidence showing that the genetic ablation of either caspsae-8 or Fadd in IECs is sufficient to induce necroptosis and spontaneous ileitis and/or colitis. Later, RIPK3 overactivation is observed in the inflamed tissues of pediatric and adult IBD patients [95]. A recent paper confirmed the positive correlation between the upregulation of necroptosis and the severity of disease in IBD patients [96].
Moreover, subjects carrying a homozygous loss-of-function mutations in RIPK1 show early-onset IBD [97].
Recently, our group showed that RIPK3-driven necroptosis seriously affects intestinal inflammation by increasing pMLKL, activating different cytokines and alarmins, and altering epithelial permeability. The inhibition of necroptosis causes a significant decrease in all these effects [98]. Accordingly, the use of the RIPK1 inhibitor necrostatin-1 (nec-1) reduces the intestinal inflammation and colitis-associated tumor growth in mice with dextran sulfate sodium ((DSS)-induced colitis [99]) while MLKL deficiency inhibits colitis by preventing inflammatory cytokines production and MAPK signaling activation [100]. MLKL-/- mice are highly susceptible to colitis and colitis-associated tumorigenesis, associated with massive leukocyte infiltration and increased inflammatory responses, suggesting a protective role of the necroptosis effector. Furthermore, the necroptosis inhibitor necrosulfonamide (NSA) represses necroptosis in intestinal epithelial cells in vitro suggesting a potential beneficial effect against IBD [101].
Lately, the expression of a secreted form of interferon lambda (IFNλ) in mice resulted in a loss of Paneth cells from intestinal tissues, via STAT1 and MLKL, controlled by caspase-8 [102].
The inhibition of HtrA2 by the serine protease UCF-101 alleviates DSS-induced colitis by preventing the necroptosis of intestinal epithelial cells [103]. Furthermore, an unexpected connection between necroptosis and the members of the disintegrin and metalloproteinase (ADAM) protease family is also described [104]. Indeed, murine embryonic fibroblasts derived from ADAM17 ex/ex mice fail to show the phosphorylation of MLKL and RIPK3 after TNF-induced necroptosis, suggesting a role of ADAM17 in this process [105].
In a recent study, Lee and Coll. [106] observe that the expression of proinflammatory cytokines in the peripheral blood mononuclear cells of UC patients and in mice with a DSS-induced colitis is markedly decreased by the administration of the RIPK3 inhibitor GSK872.
The circadian rhythm disruption (CRD) is known to be a risk factor for IBD. The genetic ablation of circadian clock function or environmental CRD in mice increases susceptibility to severe intestinal inflammation and epithelial dysregulation, accompanied by excessive necroptotic cell death and a reduced number of secretory epithelial cells, thus suggesting that CRD increases intestinal necroptosis, rendering the gut epithelium more susceptible to inflammatory processes [107].
5.2. Necrotizing Enterocolitis (NEC)
Necrotizing enterocolitis (NEC) is a rare but devastating GI disease that predominately affects preterm neonates [108]. The pathogenesis of NEC is incompletely understood and appears to be multifactorial. From a mechanistic viewpoint, it has been shown that the bacterial activation of the innate immune receptor TLR4 in the intestinal epithelium leads to barrier injury and the inflammatory microenvironment is required for the development of NEC. Indeed, the expression of TLR4 in the intestinal epithelium is higher in the premature vs. full-term intestine and mice lacking TLR4 in the intestinal epithelium are protected from NEC [109].
Recently, necroptosis has been shown to be activated in the intestinal epithelium upon TLR4 signaling and is required for NEC development [110]. The imbalance of IEC cell death, resulting in increased intestinal permeability and barrier dysfunction, leads to several acute and chronic intestinal diseases, including NEC [111].
Recently, a study shows that miR-141-3p protects IECs from LPS damage by suppressing RIPK1-mediated inflammation and necroptosis, providing an alternative perspective to explore the pathogenesis of NEC [112].
5.3. Bacterial Intestinal Infections
Infections with bacterial pathogens often results in the initiation of programmed cell death, including necroptosis, as part of the host innate immune defense, or as a bacterial virulence strategy [113].
The sexually transmitted pathogen Chlamydia trachomatis (CT) is able to replicate and survive in human IECs where induces a cytotoxic effect by various mechanisms, such as oxidative stress, apoptosis and necroptosis. Intracellular bacterial infections release DNA, sensed by DAI or TLR9, or ligands that can activate TRIF and IFN signaling that induce necroptosis, based upon the established interactions of the RHIM-containing proteins, RIPK1, RIPK3, TRIF, and DAI. Indeed, RIPK1 inhibition significantly reduces the mortality of infected cells. Authors speculate that epithelial cells, through the activation of necroptosis, can prevent the completion of pathogen replication cycles, thus blocking the progression of CT infection [114].
Moreover, Salmonella outer protein B (SopB) has a role in modulating necroptosis to facilitate the bacteria escape the epithelial cell and spread to systemic sites through a Salmonella-induced colitis model [115], while the caspase-8 knockout mice infected with Salmonella typhimurium show more severe mucosal injury and intestinal epithelial cell death as compared to wild-type mice .
Listeria monocytogenes, a bacterial foodborne pathogen, efficiently spreads and causes systemic infection in RIPK3-deficient mice while almost no dissemination is observed in wild-type mice. Intriguingly, MLKL is shown to directly bind to Listeria and inhibits their replication in the cytosol, suggesting a novel functional role of the RIPK3–MLKL pathway in nonimmune cell-derived host defense against Listeria invasion [116]. Clostridium perfringens uses its large arsenal of protein toxins to produce histotoxic, neurologic and intestinal infections in humans and animals, resulting in the activation of intracellular pathways with a variety of effects, commonly including cell death, such as apoptosis, necrosis and/or necroptosis [117]. MLKL-/- mice are more susceptible to Salmonella infection compared to their wild-type counterparts, with higher mortality rates, increased body weight loss, exacerbated intestinal inflammation, more bacterial colonization, and severe epithelial barrier disruption.
Alterations of the necroptosis key genes in intestinal human diseases are listed below (Table 1).
Table 1. Alterations of necroptotic genes in human intestinal diseases. IBD, inflammatory bowel disease; UC, ulcerative colitis; CD, Crohn’s disease; NEC, necrotizing enterocolitis; pMLKL, phospho-MLKL; pRIPK3, phospho-RIPK3; PBMC, peripheral blood mononuclear cell; nec-1, necrostatin-1.
|
Gene Alterations |
Human Model |
Effects |
Ref. |
|
RIPK3, MLKL, pMLKL upregulation |
UC inflamed biopsies |
[106] |
|
|
RIPK3 inhibition |
PBMC from UC patients treated with GSK 872 |
Necroptosis and proinflammatory cytokines reduction |
[106] |
|
pRIPK3, pRIPK1, pMLKL upregulation |
inflamed biopsies |
Increased necroptotic cell death |
[96] |
|
RIPK3 protein expression |
Colon cancer patients with metastatic stage |
High expression of RIPK3 associated with lower risk of disease progression |
[118] |
|
RIPK3, MLKL protein upregulation |
CD inflamed biopsies and serum |
Paneth cells necroptosis correlates with high level of INFλ in serum |
[102] |
|
RIPK3, MLKL protein upregulation |
NEC surgical specimen |
Increased necroptosis |
[110] |
|
loss-of-function mutations in RIPK1 |
IBD patients |
Predisposition to viral, bacterial and fungal infections, early-onset IBD, arthritis |
[97] |
|
homozygous loss-of-function mutations in RIPK1 |
Skin fibroblasts from IBD patients stimulated with TNFa or poly(I:C) |
Increased necroptosis |
[97] |
|
RIPK3 and pMLKL protein upregulation |
Inflamed biopsies from IBD pediatric patients |
Increased epithelial permeability, cytokine and alarmin expression |
[98] |
|
RIPK1 inhibition |
Colonic tissue culture from pediatric CD treated with nec-1 |
Proinflammatory cytokines reduction |
[98] |
|
RIPK3, MLKL protein upregulation |
Pediatric IBD inflamed biopsies |
[95] |
|
|
RIPK3 upregulation |
CD inflamed biopsies |
Loss of Paneth cells |
[62] |
References
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Rev. Mol. Cell. Biol. 2014, 15, 135–147, doi:10.1038/nrm3737.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrewset, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Death Differ. 2018, 25, 486–541, doi:10.1038/s41418-017-0012-4.
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. Neuroinflammation 2018, 15(1), 199, doi:10.1186/s12974-018-1235-0.
- Chan, F.K.-M.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Rev. Immunol. 2015, 33, 79–106, doi:10.1146/annurev-immunol-032414-112248.
- Orozco, S.; Oberst, A. RIPK3 in cell death and inflammation: The good, the bad, and the ugly. Rev. 2017, 277, 102–112, doi:10.1111/imr.12536.
- Christofferson, D.E.; Li, Y.; Yuan, J. Control of life-or-death decisions by RIP1 kinase. Rev. Physiol. 2014, 76, 129–150, doi:10.1146/annurev-physiol-021113-170259.
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Natl. Acad. Sci. USA 2012, 109, 5322–5327, doi:10.1073/pnas.1200012109.
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome As a Potential Therapeutic Target. Cells 2019, 8, 1486, doi:10.3390/cells8121486.
- Molnár, T.; Mázló, A.; Tslaf, V.; Szöllősi, A.G.; Emri, G.; Koncz, G. Current translational potential and underlying molecular mechanisms of necroptosis. Death Dis. 2019, 10, 860, doi:10.1038/s41419-019-2094-z.
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340, doi:10.1101/gad.312561.118.
- Liu, C.; Zhang, K.; Shen, H.; Yao, X.; Sun, Q.; Chen, G. Necroptosis: A novel manner of cell death, associated with stroke (Review). J. Mol. Med. 2018, 41, 624–630, doi:10.3892/ijmm.2017.3279.
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Rev. Pathol. 2017, 12, 103–130, doi:10.1146/annurev-pathol-052016-100247.
- Yang, C.; Li, J.; Yu, L.; Zhang, Z.; Xu, F.; Jiang, L.; Zhou, X.; He, S.; Regulation of RIP3 by the Transcription Factor Sp1 and the Epigenetic Regulator UHRF1 Modulates Cancer Cell Necroptosis. Death Dis. 2017, 8, e3084, doi:10.1038/cddis.2017.483.
- Höckendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91, doi:10.1016/j.ccell.2016.06.002.
- McCormick, K.D.; Ghosh, A.; Trivedi, S.; Wang, L.; Coyne, C.B.; Ferris, R.L.; Sarkar, S.N. Innate Immune Signaling Through Differential RIPK1 Expression Promote Tumor Progression in Head and Neck Squamous Cell Carcinoma. Carcinogenesis 2016, 37, 522–529, doi:10.1093/carcin/bgw032.
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour cell-induced Endothelial Cell Necroptosis via Death Receptor 6 Promotes Metastasis. Nature 2016, 536, 215–218, doi:10.1038/nature19076.
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting Protein Kinase 3 Is a Predictor of Survival and Plays a Tumor Suppressive Role in Colorectal Cancer. Neoplasma 2015, 62, 592–601, doi:10.4149/neo_2015_071.
- Koo, G.-B.; Morgan, M.J., Lee, D.-J.; Kim, W.J.; Yoon, J.-H.; Koo, J.S.; Kim, S.II; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent Loss of RIP3 Expression in Cancer Represses Programmed Necrosis in Response to Chemotherapeutics. Cell Res. 2015, 25, 707–725, doi:10.1038/cr.2015.56.
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. J. Pathol. 2020, 190, 272–285, doi:10.1016/j.ajpath.2019.10.012.
- Gupta, K.; Phan, N.; Wang, Q.; Liu, B. Necroptosis in cardiovascular disease—a new therapeutic target. Mol. Cell. Cardiol. 2018, 118, 26–35, doi:10.1016/j.yjmcc.2018.03.003.
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. Necroptosis is involved in CD4+T cell-mediated microvascular endothelial cell death and chronic cardiac allograft rejection. Transplantation 2017, 101, 2026–2037, doi:10.1097/TP.0000000000001578.
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.-X.; Jevnikar, A.M. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. J. Transplant. 2013, 13, 2805–2818, doi:10.1111/ajt.12447.
- Nichols, D.B.; De Martini, W.; Cottrell, J. Poxviruses Utilize Multiple Strategies to Inhibit Apoptosis. Viruses 2017, 9, 215, doi:10.3390/v9080215.
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313, doi:10.1016/j.chom.2010.03.006.
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123, doi:10.1016/j.cell.2009.05.037.
- Benedict, C.A.; Norris, P.S.; Prigozy, T.I.; Bodmer, J.L.; Mahr, J.A.; Garnett, C.T.; Martinon, F.; Tschopp, J.; Gooding, L.R.; Ware, C.F. Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2. Biol. Chem. 2001, 276, 3270–3278, doi:10.1074/jbc.M008218200.
- Jerome, K.R.; Fox, R.; Chen, Z.; Sears, A.E.; Lee, H.; Corey, L. Herpes simplex virus inhibits apoptosis through the action of two genes, Us5 and Us3. Virol. 1999, 73, 8950–8957, doi:10.1128/JVI.73.11.8950-8957.1999.
- Chen, I.L.; Tsau, J.S.; Molkentin, J.D.; Komatsu, M.; Hedrick, S.M. Mechanisms of necroptosis in T cells. Exp. Med. 2011, 208, 633–641, doi:10.1084/jem.20110251.
- Lu, J.V.; Weist, B.M.; van Raam, B.J.; Marro, B.S.; Nguyen, L.V.; Srinivas, P.; Bell, B.D.; Luhrs, K.A.; Lane, T.E.; Salvesen, G.S. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Natl. Acad. Sci. USA 2011, 108, 15312–15317, doi:10.1073/pnas.1102779108.
- Lin, J.; Chen, M.; Liu, D.; Guo, R.; Lin, K.; Deng, H.; Zhi, X.; Zhang, W.; Feng, J.; Wu, W. Exogenous hydrogen sulfide protects human umbilical vein endothelial cells against high glucose induced injury by inhibiting the necroptosis pathway. J. Mol. Med. 2018, 41, 1477–1486, doi:10.3892/ijmm.2017.3330.
- Liang, W.; Chen, M.; Zheng, D.; He, J.; Song, M.; Mo, L., Feng, J.; Lan, J. A novel damage mechanism: Contribution of the interaction between necroptosis and ROS to high glucose-induced injury and inflammation in H9c2 cardiac cells. J. Mol. Med. 2017, 40, 201–208, doi:10.3892/ijmm.2017.3006.
- Yang, X.-S.; Yi, T.-L.; Zhang, S.; Xu, Z.-W.; Yu, Z.-Q.; Sun, H.-T.; Yang, C.; Tu, Y.; Cheng, S.-X. Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Rep. 2017, 7, 5818, doi:10.1038/s41598-017-06088-0.
- Zhou, Y.; Zhou, B.; Tu, H.; Tang, Y.; Xu, C.; Chen, Y.; Zhao, Z.; Miao, Z. The degradation of mixed lineage kinase domain-like protein promotes neuroprotection after ischemic brain injury. Oncotarget 2017, 8, 68393–68401, doi:10.18632/oncotarget.19416.
- Huang, C.-Y.; Kuo, W.-T.; Huang, Y.-C.; Lee, T.-C.; Yu, L.C.H. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622, doi:10.1038/cddis.2013.149.
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195, doi:10.1038/cdd.2017.65.
- Kearney, C.J.; Martin, S.J. An inflammatory perspective on necroptosis. Cell 2017, 65, 965–973, doi:10.1016/j.molcel.2017.02.024.
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320, doi:10.1038/nature14191.
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Rev. Mol. Cell. Biol. 2010, 11, 700–715.
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Cell. Oncol. 2015, 2, e975093, doi:10.4161/23723556.2014.975093.
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Cell 2011, 43,432–448, doi:10.1016/j.molcel.2011.06.006.
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Cell 2011, 43, 449–463, doi:10.1016/j.molcel.2011.06.011.
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367, doi:10.1038/nature09852.
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. J. Mol. Sci. 2015, 16, 30321–30341, doi:10.3390/ijms161226232.
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Commun. 2017, 8, 14329, doi:10.1038/ncomms14329.
- Zhang, J.; Yang, Y.; He, W.; Sun, L. Necrosome core machinery: MLKL. Mol Life Sci. 2016, 73, 2153–2163, doi:10.1007/s00018-016-2190-5.
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Rep. 2014, 7,971–981, doi:10.1016/j.celrep.2014.04.026.
- Cai, Z.; Jitkaew,S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell. Biol. 2014, 16, 55–65, doi:10.1038/ncb2883.
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350, doi:10.1016/j.cell.2012.06.019.
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell 2012, 148, 228–243, doi:10.1016/j.cell.2011.11.030.
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, Y. et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Med. 2016, 22, 175–182, doi:10.1038/nm.4017.
- Lafont, E.; Draber, P.; Rieser, E.; Reichert, M.; Kupka, S.; de Miguel, D.; Draberova, H.; von Mässenhausen, A.; Bhamra, A.; Henderson, S.; et al. TBK1 and IKKε prevent TNF induced cell death by RIPK1 phosphorylation. Cell Biol. 2018, 20, 1389–1399, doi:10.1038/s41556-018-0229-6.
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477-1491.e19, doi:10.1016/j.cell.2018.07.041.
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Commun. 2017, 8, 359, doi:10.1038/s41467-017-00406-w.
- Seo, J.; Lee, -W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.-K.; Kim, J.-H.; Han, S.Y.; et al. CHIP controls necroptosis through ubiquitylation- and lysosome dependent degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302, doi:10.1038/ncb3314.
- Kattah, M.G.; Shao, L.; Rosli, Y.Y.; Shimizu, H.; Whang, M.I.; Advincula, R.; Achacoso, P.; Shah, S.; Duong, B.H.; Onizawa, M.; et al. A20 and ABIN-1 synergistically preserve intestinal epithelial cell survival. Exp. Med. 2018, 215, 1839–1852, doi:10.1084/jem.20180198.
- Wang, R.; Li, ; Wu, J.; Cai, Z.-Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.-H.; et al. Gut Stem Cell Necroptosis by Genome Instability Triggers Bowel Inflammation. Nature 2020, 580, 386–390, doi:10.1038/s41586-020-2127-x.
- Aizawa, S., Brar, G.; Tsukamoto, H. Cell Death and Liver Disease. Gut Liver 2020, 14,20-29, doi:10.5009/gnl18486.
- Chan, F.K.-M.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M., Orenstein, J.; Moss, B.; Lenardo, M.J. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. Biol. Chem. 2003, 278, 51613–51621, doi:10.1074/jbc.M305633200.
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840.
- Newton, K.; Wickliffe, E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 Determines Plasticity Between Cell Death Pathways. Nature 2019, 575, 679–682, doi:10.1038/s41586-019-1752-8.
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 Is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature 2019, 575, 683–687, doi:10.1038/s41586-019-1770-6.
- Günther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 2011, 77, 335–339, doi:10.1038/nature10400.
- Welz, -S.; Wullaert, A.; Vlantis, K.; Kondylis, V.; Fernández-Majada, V.; Ermolaeva, M.; Kirsch, P.; Sterner-Kock, A.; van Loo, G.; Pasparakis, M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011, 477, 330–334, doi:10.1038/nature10273.
- Hefele, M.; Stolzer, I.; Ruder, B.; He, G.-W.; Mahapatro, M.; Wirtz, S.; Neurath, M.F.; Günther, C. Intestinal Epithelial Caspase-8 Signaling Is Essential to Prevent Necroptosis During Salmonella Typhimurium Induced Enteritis. Mucosal Immunol. 2018, 11,1191-1202, doi:10.1038/s41385-018-0011-x.
- Günther, C.; Buchen, B.; He, G.-W.; Hornef, M.; Torow, N.; Neumann, H.; Wittkopf, N.; Martini, E.; Basic, M.; Bleich, A.; et al. Caspase-8 Controls the Gut Response to Microbial Challenges by Tnf-α-dependent and Independent Pathways. Gut 2015, 64, 601–610, doi:10.1136/gutjnl-2014-307226.
- Schwarzer, R.; Jiao, H.; Wachsmuth, L.; Tresch, A.; Pasparakis, M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- And GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 2020, 52, 978–993.e6, doi:10.1016/j.immuni.2020.04.002.
- Patankar, J.V.; Becker, C. Cell death in the gut epithelium and implications for chronic inflammation. Rev. Gastroenterol. Hepatol. 2020, 17, 543–556, doi:10.1038/s41575-020-0326-4.
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Commun. 2015, 6, 7515, doi:10.1038/ncomms8515.
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Commun. 2015, 6, 6282, doi:10.1038/ncomms7282.
- Yu, S.X.; Chen, W.; Liu, Z.Z.; Zhou, F.H.; Yan, S.Q.; Hu, G.Q.; Qin, X.X.; Zhang, J.; Ma, K.; Du, C.T.; et al. Non-Hematopoietic MLKL Protects Against Salmonella Mucosal Infection by Enhancing Inflammasome Activation. Immunol. 2018, 9, 119, doi:10.3389/fimmu.2018.00119.
- Shindo, R.; Ohmuraya, M.; Komazawa-Sakon, S.; Miyake, S.; Deguchi, Y.; Yamazaki, S.; Nishina, T.; Yoshimoto, T.; Kakuta, S.; Koike, M.; et al. Necroptosis of Intestinal Epithelial Cells Induces Type 3 Innate Lymphoid Cell-Dependent Lethal Ileitis. iScience 2019, 15, 536–551, doi:10.1016/j.isci.2019.05.011.
- Baxt, L.A.; Xavier, R.J. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology 2015, 149, 553–562, doi:10.1053/j.gastro.2015.06.046.
- Lalaoui, N.; Lindqvist, L.M.; Sandow, J.J.; Ekert, P.G. The molecular relationships between apoptosis, autophagy and necroptosis. Cell Dev. Biol. 2015, 39,63–69, doi:10.1016/j.semcdb.2015.02.003.
- D’Arcy, M.S. Cell death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592, doi:10.1002/cbin.11137.
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Cell 2016, 37, 337–349, doi:10.1016/j.devcel.2016.04.018.
- Matsuzawa‑Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard‑Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy Protein ATG16L1 Prevents Necroptosis in the Intestinal Epithelium. Exp. Med. 2017, 214, 3687–3705, doi:10.1084/jem.20170558.
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschenthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Häsler, R.; et al. ATG16L1 Orchestrates interleukin-22 Signaling in the Intestinal Epithelium via cGAS–STING. Exp. Med. 2018, 215, 2868–2886, doi:10.1084/jem.20171029.
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y. Gut Epithelial TSC1/mTOR Controls RIPK3-dependent Necroptosis in Intestinal Inflammation and Cancer. Clin. Invest. 2020, 130, 2111–2128, doi:10.1172/JCI133264.
- Roy, ; Esmaeilniakooshkghazi, A.; Patnaik, S.; Wang, Y.; George, S.P.; Ahrorov, A.; Hou, J.K.; Herron, A.J.; Sesaki, H.; Khurana, S. Villin-1 and Gelsolin Regulate Changes in Actin Dynamics That Affect Cell Survival Signaling Pathways and Intestinal Inflammation. Gastroenterology 2018, 154, 1405–1420.e2, doi:10.1053/j.gastro.2017.12.016.
- Lin, S.Y.; Hsieh, S.Y.; Fan, Y.T.; Wei, W.C.; Hsiao, P.W.; Tsai, D.H.; Wu, T.S.; Yang, N.S. Necroptosis Promotes Autophagy-Dependent Upregulation of DAMP and Results in Immunosurveillance. Autophagy 2018, 14, 778–795, doi:10.1080/15548627.2017.1386359.
- Otsubo, K.; Maeyashiki, C.; Nibe, Y.; Tamura, A.; Aonuma, E.; Matsuda, H.; Kobayashi, M.; Onizawa, M.; Nemoto, Y.; Nagaishi, T.; et al. Receptor-Interacting Protein Kinase 3 (RIPK3) Inhibits Autophagic Flux During Necroptosis in Intestinal Epithelial Cells. FEBS Lett. 2020, 594, 1586–1595, doi:10.1002/1873-3468.13748.
- Frank, D.; Vaux, D.L.; Murphy, J.M.; Vince, J.E.; Lindqvist, L.M. Activated MLKL Attenuates Autophagy Following Its Translocation to Intracellular Membranes. Cell Sci. 2019, 132, jcs220996, doi:10.1242/jcs.220996.
- Wallach, D.; Kang, T.B.; Kovalenko, A. Concepts of tissue injury and cell death in inflammation: A historical perspective. Rev. Immunol. 2014, 14, 51–59, doi:10.1038/nri3561.
- Orozco, S.L.; Daniels, B.P.; Yatim, N.; Messmer, M.N.; Quarato, G.; Chen-Harris, H.; Cullen, S.P.; Snyder, A.G.; Ralli-Jain, P.; Frase, S.; et al. RIPK3 Activation Leads to Cytokine Synthesis that Continues after Loss of Cell Membrane Integrity. Cell Rep. 2019, 28, 2275–2287, doi:10.1016/j.celrep.2019.07.077.
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615, doi:10.1111/febs.13775.
- Newton, K.; Manning, G. Necroptosis and Inflammation. Rev. Biochem. 2016, 85, 743–763, doi:10.1146/annurev-biochem-060815-014830.
- Moriwaki, K.; Balaji, S.; McQuade, T.; Malhotra, N.; Kang, J.; Chan, F.K.-M. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 2014, 41, 567–578, doi:10.1016/j.immuni.2014.09.016.
- Lee, K.-H.; Kang, T.-B. The Molecular Links between Cell Death and Inflammasome. Cells 2019, 8, 1057, doi:10.3390/cells8091057.
- Polykratis, A.; Martens, A.; Eren, R.O.; Shirasaki, Y.; Yamagishi, M.; Yamaguchi, Y.; Uemura, S.; Miura, M.; Holzmann, B.; Kollias, G.; et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Cell Biol. 2019, 21, 731–742, doi:10.1038/s41556-019-0324-3.
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K.-M. A RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. Immunol. 2015, 194, 1938–1944, doi:10.4049/jimmunol.1402167.
- Zhao, Q.; Yu, X.; Li, M.; Liu, Y.; Han, Y.; Zhang, X.; Li, X.M.; Wu, X.; Qin, J.; Fang, J.; et al. MLKL attenuates colon inflammation and colitis-tumorigenesis via suppression of inflammatory responses. Cancer Lett. 2019, 459, 100–111, doi:10.1016/j.canlet.2019.05.034.
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400, doi:10.18632/oncotarget.10135.
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576, doi:10.1038/cdd.2016.46.
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539, doi:10.1038/s41586-020-2025-2.
- Pierdomenico, M.; Negroni, A.; Stronati, L.; Vitali, R.; Prete, E.; Bertin. ; Gough, P.J.; Aloi, M.; Cucchiara, S. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am. J. Gastroenterol. 2014, 109, 279–287, doi:10.1038/ajg.2013.403.
- Zhou, M.; He, J.; Shi, Y.; Liu, X.; Luo, S.; Cheng, C.; Ge, W.; Qu, C.; Du, P.; Chen, Y. ABIN3 negatively regulates necroptosis-induced intestinal inflammation through recruiting A20 and restricting the ubiquitination of RIPK3 in inflammatory bowel disease. J Crohns Colitis. 2020, Jun 30:jjaa131, doi:10.1093/ecco-jcc/jjaa131.
- Cuchet-Lourenco, D.; Eletto, D.; Wu, C.; Plagnol, V.; Papapietro, O.; Curtis, J.; Ceron-Gutierrez, L.; Bacon, C.M.; Hackett, S.; Alsaleem, B. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science. 2018, 361, 810–813, doi:10.1126/science.aar2641.
- Negroni, A.; Colantoni, E.; Pierdomenico, M.; Palone, F.; Costanzo, M.; Oliva, S.; Tiberti, A.; Cucchiara, S.; Stronati, L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Liver Dis. 2017, 49, 1201–1210, doi:10.1016/j.dld.2017.08.017.
- Liu, Z.Y.; Wu, B.; Guo, Y.S.; Zhou, Y.H.; Fu, Z.G.; Xu, B.Q.; Li, J.H.; Jing, L.; Jiang, J.L.; Tang, J.; et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. J. Cancer Res. 2015, 5, 3174–3185.
- Zhang, J.; Qin, D.; Yang, Y.-J.; Hu, G.-Q.; Qin, X.-X.; Du, C.T.; Chen, W. MLKL Deficiency Inhibits DSS-induced Colitis Independent of Intestinal Microbiota. Immunol. 2019, 107, 132–141, doi:10.1016/j.molimm.2019.01.018.
- Dong, W.; Zhang, M.; Zhu, Y.; Chen, Y.; Zhao, X.; Li, R.; Zhang, L.; Ye, Z.; Liang, X. Protective effect of NSA on intestinal epithelial cells in a necroptosis model. Oncotarget 2017, 8, 86726–86735, doi:10.18632/oncotarget.21418.
- Günther, C.; Ruder, B.; Stolzer, I.; Dorner, H.; He, G.W.; Chiriac, M.T.; Aden, K.; Strigli, A.; Bittel, M.; Zeissig, S.; et al. Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients With Crohn’s Disease. Gastroenterology. 2019,157,1310-1322, doi:10.1053/j.gastro.2019.07.031.
- Zhang, C.; He, A., Liu, S.; He, Q.; Luo, Y.; He, Z.; Chen, Y.; Tao, A.; Yan, J. Inhibition of HtrA2 alleviated dextran sulfate sodium (DSS)-induced colitis by preventing necroptosis of intestinal epithelial cells. Cell death and Disease. 2019, 10, 344, doi:10.1038/s41419-019-1580-7.
- Heib, M.; Rose-John, S.; Adam, D. Necroptosis, ADAM proteases and intestinal (dys)function. Rev. Cell. Mol. Biol. 2020, 353, 83–152, doi.10.1016/bs.ircmb.2020.02.001.
- Fuchslocher Chico, J.; Falk-Paulsen, M.; Luzius, A.; Saggau, C.; Ruder, B.; Bolik, J.; Schmidt-Arras, D.; Linkermann, A.; Becker, C.; Rosenstiel, P.; et al. The enhanced susceptibility of ADAM-17 hypomorphic mice to DSS-induced colitis is not ameliorated by loss of RIPK3, revealing an unexpected function of ADAM-17 in necroptosis. 2018, 9, 12941–12958, doi:10.18632/oncotarget.24410.
- Lee, S.H.; Kwon, J.Y.; Moon, J.; Choi, J.; Jhun, J.; Jung, K.; Cho, K.H.; Darlami, O.; Lee, H.H.; Jung, E.S.; et al. Inhibition of RIPK3 Pathway Attenuates Intestinal Inflammation and Cell Death of Inflammatory Bowel Disease and Suppresses Necroptosis in Peripheral Mononuclear Cells of Ulcerative Colitis Patients. Immune Netw. 2020, 20,e16, doi:10.4110/in.2020.20.e16.
- Pagel, R.; Bär, F.; Schröder, T.; Sünderhauf, A.; Künstner, A.; Ibrahim, S.M.; Autenrieth, S.E.; Kalies, K.; König, P.; Tsang, A.H.; et al. Circadian rhythm disruption impairs tissue homeostasis and exacerbates chronic inflammation in the intestine. FASEB J. 2017, 31, 4707–4719, doi:10.1096/fj.201700141RR.
- Wang, K.; Tao, G.; Sun, Z.; Sylvester, K.G. Recent Potential Noninvasive Biomarkers in Necrotizing Enterocolitis. Res. Pract. 2019;8413698, doi:10.1155/2019/8413698.
- Leaphart, C.L.; Cavallo, J.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol. 2007,179,4808-20, doi:10.4049/jimmunol.179.7.4808.
- Werts, A.D.; Fulton, W.B.; Ladd, M.R.; Saad-Eldin, A.; Chen, Y.X.; Kovler, M.L.; Jia, H.; Banfield, E.C.; Buck, R.H.; Goehring, K.; et al. Novel Role for Necroptosis in the Pathogenesis of Necrotizing Enterocolitis. Mol. Gastroenterol. Hepatol. 2020;9,403-423, doi:10.1016/j.jcmgh.2019.11.002.
- Subramanian, S.; Geng, H.; Tan, X.D. Cell death of intestinal epithelial cells in intestinal diseases. Sheng Li Xue Bao 2020, 72, 308–324.
- Li, X.; Wang, Y.; Wang, Y.; He, X. MiR-141-3p ameliorates RIPK1-mediated necroptosis of intestinal epithelial cells in necrotizing enterocolitis. Aging (Albany NY). 2020 Jul 23;12, doi:10.18632/aging.103608.
- Demarco, B.; Chen, K.W.; Broz, P. Cross talk between intracellular pathogens and cell death. Rev. 2020, 297,174-193, doi:10.1111/imr.12892.
- Foschi, C.; Bortolotti, M.; Marziali, G.; Polito. ; Marangoni, A.; Bolognesi, A. Survival and death of intestinal cells infected by Chlamydia trachomatis. PLoS ONE. 2019, 14, e0215956, doi:10.1371/journal.pone.0215956.
- Hu, G.Q.; Yang, Y.J.; Qin, X.X.; Qi, S.; Zhang, J.; Yu, S.X.; Du, C.T.; Chen, W. Salmonella Outer Protein B Suppresses Colitis Development via Protecting Cell from Necroptosis. Cell Infect. Microbiol. 2019, 9, 87, doi:10.3389/fcimb.2019.00087.
- Sai, K.; Parsons, C.; House, J.S.; Kathariou, S.; Ninomiya-Tsuji, J. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing Cell Biol. 2019, 218, 1994–2005, doi:10.1083/jcb.201810014.
- Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of Action and Cell Death Associated with Clostridium perfringens 2018, 10, 212, doi:10.3390/toxins10050212.
- Zhang, J.; Qin, D.; Yang, Y.-J.; Hu, G.-Q.; Qin, X.-X.; Du, C.T.; Chen, W. MLKL Deficiency Inhibits DSS-induced Colitis Independent of Intestinal Microbiota. Mol. Immunol. 2019, 107, 132–141, doi:10.1016/j.molimm.2019.01.018.


