 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sara Feola | + 4373 word(s) | 4373 | 2020-10-03 02:46:42 | | | |
| 2 | Bruce Ren | Meta information modification | 4373 | 2020-10-14 04:02:30 | | | | |
| 3 | Bruce Ren | Meta information modification | 4373 | 2020-10-26 09:40:09 | | |
Video Upload Options
According to the latest available data, cancer is the second leading cause of death, highlighting the need for novel cancer therapeutic approaches. In this context, immunotherapy is emerging as a reliable first-line treatment for many cancers, particularly metastatic melanoma. Indeed, cancer immunotherapy has attracted great interest following the recent clinical approval of antibodies targeting immune checkpoint molecules, such as PD-1, PD-L1, and CTLA-4, that release the brakes of the immune system, thus reviving a field otherwise poorly explored. Cancer immunotherapy mainly relies on the generation and stimulation of cytotoxic CD8 T lymphocytes (CTLs) within the tumor microenvironment (TME), priming T cells and establishing efficient and durable anti-tumor immunity. Therefore, there is a clear need to define and identify immunogenic T cell epitopes to use in therapeutic cancer vaccines. Naturally presented antigens in the human leucocyte antigen-1 (HLA-I) complex on the tumor surface are the main protagonists in evocating a specific anti-tumor CD8+ T cell response. However, the methodologies for their identification have been a major bottleneck for their reliable characterization. Consequently, the field of antigen discovery has yet to improve.
1. Introduction
The recent clinical success of antibodies targeting immune checkpoint molecules, such as programmed death receptor-1 (PD-1), its ligand PD-L1, and cytotoxic T cell-associated antigen 4 (CTL-A4), have led to a new and strong interest in the field of cancer immunotherapy [1][2]. Immune checkpoint inhibitors (ICIs) release the brakes of the immune system, reviving and boosting the effector function of specific anti-tumor T cells [3]. In 2018, James Patrick Allison and Tasuku Honjo received the Nobel prize for medicine “for their discovery of cancer therapy by inhibition of negative immune regulation” [4][5]. The overall response to ICIs is reportedly unsatisfactory for many types of cancer [6], highlighting the need to combine ICIs with cancer immunotherapeutic approaches, such as therapeutic cancer vaccines, with the artificial generation, stimulation, and tumor microenvironment (TME) infiltration of cancer-specific CD8+ T cells called cytotoxic T lymphocytes (CTLs) [7][8][9]. CTLs patrol the whole organism, checking the major histocompatibility complex (MHC), called the human leucocytes antigen (HLA) in humans, that possesses an antigenic peptide that is typically 8–11 aminoacidic residues in length and expressed on the cellular surface. If an aberrant peptide is eventually spotted in the HLA-I complex, the CTLs kill the cell.
In this context, to create effective tumor protection and rejection strategies, the reliable identification of tumor antigens in the HLA-I complex plays a pivotal role. To date, the methodologies for the correct identification of antigens in the HLA-I complex rely on the direct isolation of peptides from the HLA-I complex and/or the in silico prediction of relevant antigens [10][11].
The direct identification of peptides from the HLA complex is accomplished using different techniques, of which immunoaffinity purification (HLA immunoprecipitation) and the extraction of HLA peptides are the most well-established [12]. Isolated peptides are identified by tandem mass-spectrometry (MS/MS). The entire process is highly laborious and time-consuming [13][14]. It has been reported that immunoaffinity purification produces a low yield (i.e., 1–3%) [15], even though additional experiments in different settings are needed to better determine how much of the sample is lost [16]. The peptide isolation procedure is universally recognized as the bottleneck of the entire procedure and therefore requires improvement [16].
In the antigen discovery process, in silico tools are used to predict immunologically relevant antigens. The identification of target candidates relies on bioinformatic approaches that allow a structural analysis of genomic and proteomic data. For instance, next generation sequencing (NGS) approaches can benefit from the use of epitope prediction tools to combine two kinds of information: the differential gene expression in cancer compared to matched healthy tissue and the probability of those candidates to be presented on the cell surface onto the HLA molecule [17][18]. To achieve this, the actual bioinformatic tools contain T cell epitope algorithms that are able to predict putative T cell epitopes based on the well-characterized rules to which HLA-I presented peptides adhere [19][20]. Indeed, several predictors of HLA binders have been developed [21]; these will be defined in Section 4. HLA binding is only one part of the story; today, the entire processing machinery can be taken into account (e.g., proteasomal cleavage, transporter-associated antigen processing (TAP) transport) by the bioinformatic algorithms to predict relevant T cell epitopes [21].
NGS and in silico tools are effective methods in antigen discovery; however, improvements are needed. These methods lack rich and experimentally validated datasets, thus decreasing the accuracy of the predictive algorithms. The bioinformatic predictions give hints, but the selection of relevant epitopes from among the candidates always requires experimental validation [21].
2. Antigens
The word “antigen” refers to the molecular structure seen by the antibodies or to any molecule or linear molecular fragment derived from the processing of the native antigen that can be recognized by T cell receptors (TCRs) [22]. This paper mainly examines CD8+ T cell-restricted antigens. The existence and role of tumor antigens have been discussed since 1940 and were reported on before the discovery of T cells, which occurred in the 1960s[23][24]. In 1943, Gross et al. performed a pivotal experiment that showed for the first time the role of the immune system in tumor rejection [25]. Briefly, mice were treated with methylcholanthrene and subsequently developed tumors. The tumors were then resected, and the tumor cells were implanted back into the mice. The mice then rejected the second tumor. This experiment demonstrated that acquired immunity was induced and directed against the tumor and that the rejection did not depend upon genetic differences between the inoculated mice and the mice that produced the tumor cells [25].
Later, Boon and colleagues achieved similar results using mutagen-treated murine cell lines that failed to form tumors in syngeneic mice; this research confirmed that tumors express antigens recognized by CTLs and confirmed the role of the immune system in rejecting malignant cells [26][27]. However, the molecular nature of the antigens expressed by tumors and recognized by CTLs was only discovered in 1989 by Lurquin et al. [28]. The researchers identified a single peptide recognized by CTLs that differed from the self-protein by single point mutation. This observation clearly showed that upon mutation, the tumors expressed altered proteins, thus labeling the cells for CTL recognition [28][29].
Today, based on the expression of the parental gene, tumor antigens can be classified into the general categories of tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) [30] (Figure 1).
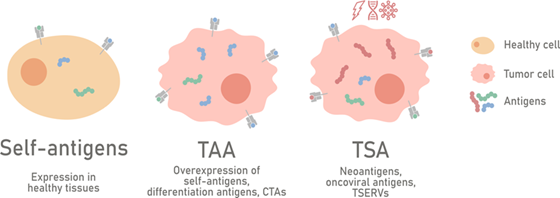
Figure 1. Classification of tumor antigens. Tumor antigens can generally be categorized into tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) based on the expression pattern of the parental gene. TAAs are self-proteins expressed in cancer cells; upon malignant transformation, the following consequences can be observed: the overexpression of normal proteins (gene overexpressed), the expression of proteins with tissue-specific gene patterns (differentiation antigens), or the expression of proteins derived from gene expression restricted to the testes (cancer germline/cancer testis antigens). TSAs are proteins expressed by tumor cells and can arise from mutations (neoantigens), from viruses involved in the oncogenic transformation (oncoviral antigens), or from the expression of tumor-specific endogenous retroviruses (TSERVs).
2.1. Tumor-Associated Antigens
TAAs are a large family of antigens that includes antigens derived from genes overexpressed in tumors, differentiation antigens, and cancer germline/cancer testis antigens [30][31]. Antigens derived from genes overexpressed in tumors comprise a class of normal self-proteins which are minimally expressed by healthy tissues but constitutively overexpressed in cancer cells as a result of their malignant profile. The proteins which are usually overexpressed (e.g., EGFR, hTERT, p53, carbonic anhydrase IX) are mainly involved in the survival of the cancer cells and therefore are not susceptible to downregulation mechanisms, making them an attractive target for cancer therapeutic approaches [31]. For instance, in 1996, Gaugler et al. discovered an overexpressed antigen in renal cell carcinoma (RCC) called renal antigen 1 (RAGE-1) [32]. RAGE-1 is the first example of an antigen recognized by autologous CTLs in RCC, and its expression is restricted to the retina and to different histological tumor types. As the retina does not express HLA-I [33], RAGE-1 becomes a possible candidate to target for cancer immunotherapy [34][35]. Another interesting example is HER2/NEU, which is overexpressed in epithelial tumors such as ovarian and breast tumors [36][37]. The use of trastuzumab, a monoclonal antibody targeting the extracellular domain of HER2, has revolutionized the treatment of breast cancer HER2+ [38]. In addition, HER2 is a suitable candidate for the peptide vaccine approach; indeed, the nonapeptide E75, derived from the HER2 sequence, has been described as being able to elicit four different ovarian CTL cell line responses [39]. E75 as a monotherapy (Nelipepimut-S) or in combination with granulocyte macrophage colony-stimulating factor (GM-CSF) (NeuVAx) has been tested in clinical trials [40][41][42]. The genes belonging to the apoptosis pathway are also upregulated in tumor cells compared to healthy tissues, representing a source of T cell epitopes. For instance, a peptide derived from survivin, an apoptosis inhibitor protein, was used to generate CTLs in vitro from healthy donors; matched cell lines and primary malignant cells from patients were then lysed by those T cells [43]. In addition, a p53 derived wild type peptide (L9V) was used to generate in vitro CTLs that were able to kill squamous carcinoma cell lines in an L9V-HLA-dependent fashion [44].
As TAAs have a higher expression level in tumors compared to normal tissue and are shared among several tumors, a safe use for them has been proposed for cancer therapeutic approaches. For instance, novel chimeric antigen receptor (CAR) T cells have largely been adopted to target this class of antigens . CAR T cells consist of an extracellular domain made of the variable region of the antibody’s heavy and light chains linked to the intracellular signaling domain (CD3-zeta, CD28, 41BB). This characteristic makes the CAR T cells able to kill the target in an HLA-independent manner [45]. The adoption of anti-CD19 CARs has showed good clinical outcomes in the treatment of B cell lymphomas and leukemias [46][47][48].
However, the main drawback in using TAAs in cancer immunotherapy is the feasibility of the expression analysis of these proteins in every single tissue under every physiological condition; this hinders a comprehensive safety profile of the TAAs . Indeed, potential hazards (i.e., “on-target, off-tumor” toxicity, onset of autoimmune disease) associated with the clinical treatment based on these molecules have been reported. For instance, CAR T cells targeting carbonic anhydrase IX (CAIX) in RCC patients induced liver toxicity, requiring cessation of the treatment. Biopsies revealed CAIX expression in the bile duct epithelium with T cell infiltration, including CAR T cells; this is a typical example of “on-target, off-tumor” toxicity [49]. Moreover, the central and peripheral tolerance mechanisms eliminate the T and B cells’ ability to recognize self-antigens. A TAA peptide-based vaccine must break the tolerance, thus stimulating the low-affinity and rare T cells still circulating. This could interfere with the development of a proper cancer therapeutic vaccine [50]. The identification and use of an effective vaccine adjuvant could overcome the problem, delivering benefits to cancer patients [51].
The differentiation antigens represent normal proteins which are expressed as a consequence of a specific function of the target tissue. These were first reported in melanoma in which proteins (e.g., tyrosinase, Melan-A/MART-1, gp100/Pmel17) involved in melanin production or melanosome generation were often observed to be targets for CTLs from melanoma patients and healthy donors [52][53]. In the 1993 work of Brichard et al., lymphocytes derived from two melanoma patients were stimulated with irradiated cells from the autologous melanoma, and CTLs that were able to lyse them were obtained. The authors then demonstrated that the lysis was antigen HLA-0201-specific, and upon using the cloning approach, they identified the gene encoding the antigen as tyrosinase [53]. Rosenberg and colleagues administered tumor-infiltrating lymphocytes (TILs) to treat metastatic melanoma patients and had good results. The authors used the TILs to clone and determine the antigens involved in the anti-tumor response. In 1994, Rosenberg and colleagues established a CTL cell line called TIL1200 from a metastatic melanoma patient and were able to lyse the autologous melanoma and other melanoma HLA-0201+ cell lines. The authors showed that TIL1200 recognized a self-antigen not mutated in the HLA-0201 context and that the antigen sequence belonged to a membrane glycoprotein known as gp100/Pmel17 [54]. In the same year, Rosenberg and colleagues identified a shared and commonly expressed HLA-0201-restricted melanoma antigen that was recognized by T cells 1 (MART-1), applying a similar approach to that used to isolate gp100/Pmel17 [55]. In the following years, other melanoma differentiation antigens were identified, such as TRP1/gp75 and TRP2 (tyrosinase-related proteins), in the context of the HLA-A31 molecule [56][57]. Later, the TRP2 case was reviewed to identify an HLA-0201-restricted peptide, as HLA-0201 occurs frequently in the population. Based on the peptide-binding motif for HLA-A0201 and experimental validation, TRP2 peptides 180–188 were reported, and the sequence was found to be identical to the one recognized by H2Kb-restricted B16 murine melanoma CTLs, paving the way for a murine tumor immunotherapy model [58]. Furthermore, differentiation antigens such as prostatic acid phosphatase (PAP) and prostate-specific antigen (PSA) are used for prostate cancer immunotherapy. Naturally expressed HLA-02 restricted peptides from PAP were identified through sequence analysis and in vitro validation [59]. Moreover, two PSA HLA-02-restricted peptides which are capable of eliciting CTL responses have been reported by Correale et al. [60]. In addition, immunization experiments using HLA-0201 transgenic mice were employed to identify a peptide from carcinoembryonic antigens (CEA) [61]. This is a glycoprotein overexpressed in colon rectal cancer and other selected epithelial cancers, while in normal conditions, its expression is restricted to fetal development and in adults’ tissue in the epithelial cells of the gastrointestinal tract [62][63].
The main disadvantage in exploiting differentiation antigens as vaccines for cancer therapy is the onset of autoimmune toxicity. In cases of targeting melanoma-melanocyte antigens, reactions such as severe skin rashes and vitiligo lesions have been reported [64][65]. CAR T cells targeting CEA that were used to treat three patients with metastatic colorectal cancer showed a promising outcome in at least one patient; however, severe transient colitis was induced, most probably due to the CEA presence in the colonic mucosa [62]. These observations highlight the importance of targeting tumor-specific antigens and/or antigens with limited expression in normal tissues (e.g., cancer germline/cancer testis antigens).
Cancer germline/cancer testis antigens (CTAs) are a large family (the CT database consists of 204 genes [66]) of tumor-associated antigens expressed in human tumors of different histological origins but not in normal tissue, except for testis and placenta tissue [67]. In 1991, Van der Bruggen and co-workers identified the first human gene that encoded for a cancer testis antigen [68]. The authors isolated the CTLs that were capable of lysing the autologous cell line derived from the melanoma patient MZ2. Using a clonal sub-line and a DNA cloning approach, they identified the gene which was able to sensitize the sub-line to the CTL lysis. This gene was named melanoma antigen family A, 1 (MAGEA1), and it is expressed in many human tumors of different histological types. No expression was detected in normal tissues, with the exception of testis and placenta tissue [68][69]. In the same work, another two MAGE members were reported, MAGE A2 and MAGE A3 [68]. Subsequently, the nonapeptides derived from HLA-A1-restricted MAGE A1 and MAGE A3 were described [70][71].
After this, many antigens were identified in diverse tumors with restricted expression in testis tissue; Old and Chen called these cancer testis (CT) antigens [72]. To date, the MAGE family is subdivided into type I and type II. Type I comprises three sub-families, MAGE-A, -B, and -C, and contains the relevant CTAs. Type II includes the MAGE-D, -E, -F, -G, -H, and -L sub-families and Necdin that are expressed in different adult tissues [73][74][75]. Several members have been identified in the MAGE group [76], and MAGE A3 is one of the most frequently expressed TAAs in many tumors, including melanoma, non-small cell lung carcinoma (NSCLC), and head and neck tumors [77]. In melanoma, the preferentially expressed antigen of melanoma (PRAME) is another example of a CTA; PRAME is defined as a testis-selective rather than a testis-restricted CTA as its expression has been observed in endometrial, ovarian, and adrenal gland tissues in addition to testis tissue [78]. Building on the work by Ikeda and co-workers [79], Kessler et al. subsequently identified four HLA-0201 peptides that were restricted in PRAME [80]. In 2011, Quintarelli and colleagues were able to generate PRAME-specific CTLs from both healthy donors and leukemia patients [81]. In 1997, the serological analysis of recombinant tumor cDNA expression libraries (SEREX) technique was used by Chen et al. to identify novel tumor antigens in esophageal squamous cell carcinoma patients; the screening revealed sequences belonging to eight genes, among them New York esophageal squamous cell carcinoma 1 (NY-ESO 1), that were expressed in normal tissue, such as testis and ovarian tissue, and in several tumors, such as melanoma, breast cancer, bladder cancer, prostate cancer, and hepatocellular carcinoma [82]. Since then, NY-ESO1 has been employed in several immunotherapy-based treatments [83]. For instance, recombinant NY-ESO1 protein was used in combination with the adjuvant ISCOMATRIX in 46 patients with resected NY-ESO1+ tumors. Overlapping peptides were then employed to verify the T cells’ response to NY-ESO1 upon vaccination, showing CD4+ and CD8+ T cells’ specific response to both known and uncharacterized peptides derived from NY-ESO1 [84]. In 1997, Türeci et al. used the SEREX technique to investigate human melanoma and described a novel CTA gene called synovial sarcoma X chromosome breakpoint (SSX2) [85], to which a T cell response has been reported in patients with tumors of diverse histological origins [86]. An analysis of the sub-clones derived from the MZ2 melanoma patient allowed the identification of the B melanoma antigen (BAGE) gene and the HLA-Cw1601-restricted peptide [87]; BAGE expression was found only in testis and tumor (e.g., bladder cancer) tissues, and thus was found to be a member of the CTA family. Using a similar experimental procedure, other CTAs were identified—the gene G antigen 1 (GAGE) and the HLACw0601-restricted peptide [88].
Being immunologically privileged sites, the testes and placenta do not express HLA molecules [89]; therefore, CTAs are promising targets for cancer immunotherapy. Indeed, MAGE A3 as a recombinant protein has been used in the largest ever phase III lung cancer clinical trial, the MAGE A3 as Adjuvant Non-Small Cell Lung Cancer Immunotherapy (MAGRIT) trial [90]. In phase II, the vaccination protocol was well tolerated and the results promising; however, MAGRIT failed to show any improvement in disease-free survival (DFS) when compared with a placebo [91]. In addition, TCR gene modified T cells to target the MAGEA3/A12 HLA-A0201 restricted peptide were used to treat nine patients with tumors expressing MAGE A3/A12 in a phase I/II clinical trial. The encouraging cancer regression that was observed was dampened by severe neurotoxicity that resulted in the deaths of two patients. Subsequent analysis revealed the expression of MAGE A12 in the brain, explaining the inflammation and neuronal degeneration that was observed [92]. The use of CTAs, such as MAGE family members, is promising; however, caution is required in further applications.
2.2. Tumor-Specific Antigens
TSAs are a category of antigens restricted to tumors and are not found in healthy cells; this is the result of malignant mutations or the expression of viral elements. Neoantigens, oncoviral antigens, and endogenous retroviral elements belong to this category [93].
Neoantigens are a subset of TSAs produced as a direct consequence of genetic alteration caused by tumor DNA mutations (e.g., non-synonymous single point mutations, frameshifts, insertions/deletions) and are patient-specific [94][95]. In 1995, Coulin et al. reported the first example of a neoantigen [96]. The authors identified the source gene of the antigen that was recognized by an autologous CTL and called it melanoma ubiquitous mutated (MUM-1). As the gene was expressed ubiquitously, the authors wondered whether a mutation had occurred in the gene to induce an anti-tumor T cell response. Actually, MUM-1 in the melanoma cells carried a single point mutation that resulted in an amino acid change in the nonapeptide HLA-B44 restricted, making it suitable for interaction with the T cells [96]. Several other neoantigens have since been identified. For instance, Rosenberg and colleagues discovered a single point mutation in the b catenin sequence in one melanoma patient that resulted in a change from a serine to a phenylalanine residue at position 37; the derived peptide (SYLDSGIHF) showed a high affinity binding for HLA-A24, the patient’s HLA allele [97]. Moreover, a single point mutation in the gene encoding CDK4 resulted in the generation of an HLA-A0201-restricted peptide that was recognized by autologous CTLs in the melanoma patient, and the mutation altered the cell cycle regulation [98]. A mutation in the CASP-8 gene that reduced the function of the protein and generated an HLA-B3503-restricted peptide that was recognized by autologous CTLs was reported in a patient with squamous cell carcinoma of the oral cavity [99].
The neoantigens in cancer immunotherapy have the undiscussed advantage of being non self-antigens and hence the T cells are not affected by central tolerance, making them highly immunogenic antigens [94]. However, their main limitation is their great variability within and between tumors. Great variability within tumors can induce negative selection, allowing the survival of the cancer cells that no longer express the neoantigens. Great variability between tumors requires characterization and the development of a specific vaccine for each patient. In addition, the mutational burden plays a pivotal role in the generation of neoepitopes. Tumors with a high mutation frequency are most likely to generate neoantigens. Tumors with a low mutation burden may be difficult to investigate in terms of identifying neoantigens and subsequent vaccine production .
Oncoviral antigens consist of proteins derived from the viruses driving the oncogenic transformation; these proteins are the source of peptides present on the cellular surface in the HLA context and recognized by T cells. As oncogenic viruses are shared by the same kinds of tumors, this class of antigen is not patient-specific . Prophylactic vaccines have been produced and mainly rely on eliciting neutralizing antibodies to prevent the virus from entering the cells. The treatment of established tumors involves targeting T cell epitopes . For instance, human papillomavirus (HPV) is associated with benign papilloma or warts and cancer of the cervix, anus, penis, and head and neck [100]. Ramos et al. isolated PBMCs from patients with HPV+ cancer and stimulated them in vitro with a mix of HLA-I-restricted and HLA-II restricted peptides, covering the E6/E7 proteins of HPV16; the authors were able to demonstrate that the patients had E6- and E7-specific T cells [101]. These have been called HPV-ST, and an on-going phase I clinical trial is evaluating the effect of HPV-ST cells in treating HPV+ tumors (NCT02379520). Another example is the development of therapeutic vaccines for cancers related to the Epstein–Barr virus (EBV) which is involved in the onset of several disorders, such as B-cell lymphoproliferative disorders and nasopharyngeal carcinoma [102][103]. In a phase I clinical trial, 16 patients with established HBV+ tumors were treated with modified vaccinia Ankara (MVA) encoding the full length of LMP2 and the C-terminal of EBNA1 proteins from EBV. A specific T cell response to LMP2 and/or EBNA1 was detected, showing the feasibility of boosting an EBV-specific immune response [104]. However, the extent of the clinical benefits is still being investigated in a phase II clinical trial (NCT01094405). Oncoviral antigens lack expression in healthy cells, making them highly tumor-specific. They are also common to patients. However, 15% of cancers have a viral etiopathology, limiting their clinical application [105][106].
Endogenous retroviral elements (ERVs) or human endogenous retroviral elements (HERVs) are fragments of genomic DNA derived from the integration of retrotranscribed retroviral RNAs that infected the germ line cells of humans’ ancestors. Over time, ERVs have been vertically transmitted and, to date, they represent 8% of the human genome. ERVs have accumulated mutations over time, losing the capability of producing competent replicative viral particles [107]. Moreover, epigenetic mechanisms (e.g., methylation) suppress most of their expression in healthy cells. For instance, in the thymus, ERVs are partially epigenetically silenced, and ERVs which are reactive to T cells do not go through complete negative selection [108]. In cancer, ERVs are induced upon malignant transformation and/or epigenetic therapy, becoming targets for cancer therapeutic approaches [109]. In 2015, Rooney at al. investigated the cytolytic activity and expression pattern associated with 66 ERVs in tumors compared to healthy tissues. Surprisingly, they found three ERVs (ERVH5, ERVH48-1, and ERVE4) with minimal or undetectable expression in normal tissue and overexpression in tumors; these have been termed tumor-specific endogenous retroviruses (TSERVs) . Interestingly, in regard to ERVE4, Rooney´s data had already been experimentally validated. For instance, following hematopoietic stem cell transplantation (HSCT), RCC patients experienced disease regression. Child and colleagues found RCC-reactive CD8+T cells derived from the donor. Using a cDNA cloning approach, the authors identified the antigen in HERV-E gene products and described an HLA-A11-restricted 10-mer peptide (ATFLGSLTWK) as the target recognized by the tumor-reactive CTLs [110]. The same authors also reported the identification of three HLA-A0201-restricted peptides derived from HERV4env that were able to elicit RCC-reactive CTL responses [111]. ERVH-5 has been reported in bladder, colorectal, head and neck, lung squamous, ovarian, stomach, and uterine cancers. ERVH48-1 is prominently expressed in bladder cancer and prostate cancer [109]. Schiavetti et al. described CTLs which were reactive against the peptide derived from HERV-K-MEL in two melanoma patients. The authors determined that the peptide sequence (MLAVISCAV) was HLA-A2-restricted, showing that the CTLs’ reactivity against the peptide occurred only in the two patients and not in the healthy donors[112]. Based on the presence of HERV-K gag proteins in the cytoplasm of primary tumor cells and on the detection of antibodies to HERV-K gag in patients with seminoma, Rakoff–Nahoum investigated the HERV-K-specific T cell-mediated immune response in the blood of those patients. The authors synthetized 15 HERV-K predicted peptides based on the HLA-I binding motif and proline-enriched region. Next, PBMCs from seminoma patients and healthy donors were screened with four pools of these peptides. The T cell reactivity was higher in at least three pools of peptides in the seminoma patients compared to the healthy donors [113].
Their high tumor specificity and expression and incomplete T cell tolerance [108] make ERVs the ideal target for cancer immunotherapeutic approaches. In addition, autologous CTLs which are able to recognize HLA-restricted peptides have been reported [113], and ERVs are common to cancer patients. This allows for off-the-shelf therapy. However, the epitopes recognized by CTLs which are found in cancer patients are still few, and the expression of different HERV families in cancer is still limited. Therefore, future proteomic analysis, especially of the thymus, and an in-depth understanding of the mechanisms involved in HLA-I presentation will shed light on the use of ERVs in cancer immunotherapy [109].
References
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812, doi:10.1038/nrc3153.
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61, doi:10.1126/science.aaa8172.
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264, doi:10.1038/nrc3239.
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465, doi:10.1084/jem.182.2.459.
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895.
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255, doi:10.1186/s13046-019-1259-z.
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168, doi:10.3389/fimmu.2019.00168.
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10, doi:10.1016/j.cell.2017.08.027.
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214, doi:10.1016/j.cell.2015.03.030.
- Trolle, T.; McMurtrey, C.P.; Sidney, J.; Bardet, W.; Osborn, S.C.; Kaever, T.; Sette, A.; Hildebrand, W.H.; Nielsen, M.; Peters, B. The Length Distribution of Class I-Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele-Specific Binding Preference. J. Immunol. 2016, 196, 1480–1487, doi:10.4049/jimmunol.1501721.
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707, doi:10.1038/s41596-019-0133-y.
- Bassani-Sternberg, M.; Coukos, G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17, doi:10.1016/j.coi.2016.04.005.
- Kowalewski, D.J.; Stevanovic, S. Biochemical large-scale identification of MHC class I ligands. Methods Mol. Biol. 2013, 960, 145–157, doi:10.1007/978-1-62703-218-6_12.
- Bassani-Sternberg, M. Mass Spectrometry Based Immunopeptidomics for the Discovery of Cancer Neoantigens. Methods Mol. Biol. 2018, 1719, 209–221, doi:10.1007/978-1-4939-7537-2_14.
- Hassan, C.; Kester, M.G.; Oudgenoeg, G.; de Ru, A.H.; Janssen, G.M.; Drijfhout, J.W.; Spaapen, R.M.; Jimenez, C.R.; Heemskerk, M.H.; Falkenburg, J.H.; et al. Accurate quantitation of MHC-bound peptides by application of isotopically labeled peptide MHC complexes. J. Proteom. 2014, 109, 240–244, doi:10.1016/j.jprot.2014.07.009.
- Caron, E.; Aebersold, R.; Banaei-Esfahani, A.; Chong, C.; Bassani-Sternberg, M. A Case for a Human Immuno-Peptidome Project Consortium. Immunity 2017, 47, 203–208, doi:10.1016/j.immuni.2017.07.010.
- Backert, L.; Kohlbacher, O. Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med. 2015, 7, 119, doi:10.1186/s13073-015-0245-0.
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421, doi:10.1172/JCI80008.
- Rotzschke, O.; Falk, K.; Stevanovic, S.; Jung, G.; Walden, P.; Rammensee, H.G. Exact prediction of a natural T cell epitope. Eur. J. Immunol. 1991, 21, 2891–2894, doi:10.1002/eji.1830211136.
- Falk, K.; Rotzschke, O.; Stevanovic, S.; Jung, G.; Rammensee, H.G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 1991, 351, 290–296, doi:10.1038/351290a0.
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inf. 2015, 53, 405–414, doi:10.1016/j.jbi.2014.11.003.
- Murphy, K.; Weaver, C. Janeway's Immunobiology, 9th ed.; Garland Science/Taylor & Francis Group, LLC: New York, NY, USA, 2016; pp. 14, 904 pages.
- Miller, J.F.; Mitchell, G.F. The thymus and the precursors of antigen reactive cells. Nature 1967, 216, 659–663, doi:10.1038/216659a0.
- Miller, J.F. Events that led to the discovery of T-cell development and function—A personal recollection. Tissue Antigens 2004, 63, 509–517, doi:10.1111/j.0001-2815.2004.00255.x.
- Gross, L. Intradermal Immunization of C3H Mice against a Sarcoma That Originated in an Animal of the Same Line. Cancer Res. 1943, 3, 326–333.
- Boon, T.; Kellermann, O. Rejection by syngeneic mice of cell variants obtained by mutagenesis of a malignant teratocarcinoma cell line. Proc. Natl. Acad. Sci. USA 1977, 74, 272–275, doi:10.1073/pnas.74.1.272.
- Van Pel, A.; Boon, T. Protection against a nonimmunogenic mouse leukemia by an immunogenic variant obtained by mutagenesis. Proc. Natl. Acad. Sci. USA 1982, 79, 4718–4722, doi:10.1073/pnas.79.15.4718.
- Lurquin, C.; Van Pel, A.; Mariame, B.; De Plaen, E.; Szikora, J.P.; Janssens, C.; Reddehase, M.J.; Lejeune, J.; Boon, T. Structure of the gene of tum- transplantation antigen P91A: The mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 1989, 58, 293–303, doi:10.1016/0092-8674(89)90844-1.
- Vigneron, N.; Van den Eynde, B.J. Insights into the processing of MHC class I ligands gained from the study of human tumor epitopes. Cell. Mol. Life Sci. 2011, 68, 1503–1520, doi:10.1007/s00018-011-0658-x.
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7, doi:10.1038/s41541-019-0103-y.
- Ilyas, S.; Yang, J.C. Landscape of Tumor Antigens in T Cell Immunotherapy. J. Immunol. 2015, 195, 5117–5122, doi:10.4049/jimmunol.1501657.
- Gaugler, B.; Brouwenstijn, N.; Vantomme, V.; Szikora, J.P.; Van der Spek, C.W.; Patard, J.J.; Boon, T.; Schrier, P.; Van den Eynde, B.J. A new gene coding for an antigen recognized by autologous cytolytic T lymphocytes on a human renal carcinoma. Immunogenetics 1996, 44, 323–330, doi:10.1007/bf02602776.
- Abi-Hanna, D.; Wakefield, D.; Watkins, S. HLA antigens in ocular tissues. I. In vivo expression in human eyes. Transplantation 1988, 45, 610–613, doi:10.1097/00007890-198803000-00021.
- Oehlrich, N.; Devitt, G.; Linnebacher, M.; Schwitalle, Y.; Grosskinski, S.; Stevanovic, S.; Zoller, M. Generation of RAGE-1 and MAGE-9 peptide-specific cytotoxic T-lymphocyte lines for transfer in patients with renal cell carcinoma. Int. J. Cancer 2005, 117, 256–264, doi:10.1002/ijc.21200.
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. Biomed. Res. Int. 2015, 2015, 948501, doi:10.1155/2015/948501.
- Kraus, M.H.; Popescu, N.C.; Amsbaugh, S.C.; King, C.R. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 1987, 6, 605–610.
- Pils, D.; Pinter, A.; Reibenwein, J.; Alfanz, A.; Horak, P.; Schmid, B.C.; Hefler, L.; Horvat, R.; Reinthaller, A.; Zeillinger, R.; et al. In ovarian cancer the prognostic influence of HER2/neu is not dependent on the CXCR4/SDF-1 signalling pathway. Br. J. Cancer 2007, 96, 485–491, doi:10.1038/sj.bjc.6603581.
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51, doi:10.1056/NEJMra043186.
- Fisk, B.; Blevins, T.L.; Wharton, J.T.; Ioannides, C.G. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J. Exp. Med. 1995, 181, 2109–2117, doi:10.1084/jem.181.6.2109.
- Mittendorf, E.A.; Lu, B.; Melisko, M.; Price Hiller, J.; Bondarenko, I.; Brunt, A.M.; Sergii, G.; Petrakova, K.; Peoples, G.E. Efficacy and Safety Analysis of Nelipepimut-S Vaccine to Prevent Breast Cancer Recurrence: A Randomized, Multicenter, Phase III Clinical Trial. Clin. Cancer Res. 2019, 25, 4248–4254, doi:10.1158/1078-0432.CCR-18-2867.
- Benavides, L.C.; Gates, J.D.; Carmichael, M.G.; Patil, R.; Holmes, J.P.; Hueman, M.T.; Mittendorf, E.A.; Craig, D.; Stojadinovic, A.; Ponniah, S.; et al. The impact of HER2/neu expression level on response to the E75 vaccine: From U.S. Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin. Cancer Res. 2009, 15, 2895–2904, doi:10.1158/1078-0432.CCR-08-1126.
- Clifton, G.T.; Peoples, G.E.; Mittendorf, E.A. The development and use of the E75 (HER2 369-377) peptide vaccine. Future Oncol. 2016, 12, 1321–1329, doi:10.2217/fon-2015-0054.
- Schmidt, S.M.; Schag, K.; Muller, M.R.; Weck, M.M.; Appel, S.; Kanz, L.; Grunebach, F.; Brossart, P. Survivin is a shared tumor-associated antigen expressed in a broad variety of malignancies and recognized by specific cytotoxic T cells. Blood 2003, 102, 571–576, doi:10.1182/blood-2002-08-2554.
- Ropke, M.; Hald, J.; Guldberg, P.; Zeuthen, J.; Norgaard, L.; Fugger, L.; Svejgaard, A.; Van der Burg, S.; Nijman, H.W.; Melief, C.J.; et al. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53-derived peptide. Proc. Natl. Acad. Sci. USA 1996, 93, 14704–14707, doi:10.1073/pnas.93.25.14704.
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68, doi:10.1126/science.aaa4967.
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720, doi:10.1182/blood-2011-10-384388.
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023, doi:10.1182/blood-2014-12-580068.
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102, doi:10.1182/blood-2010-04-281931.
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912, doi:10.1038/mt.2013.17.
- Pedersen, S.R.; Sorensen, M.R.; Buus, S.; Christensen, J.P.; Thomsen, A.R. Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: Implications for cancer immunotherapy. J. Immunol. 2013, 191, 3955–3967, doi:10.4049/jimmunol.1300555.
- Overwijk, W.W. Cancer vaccines in the era of checkpoint blockade: The magic is in the adjuvant. Curr. Opin. Immunol. 2017, 47, 103–109, doi:10.1016/j.coi.2017.07.015.
- Pittet, M.J.; Valmori, D.; Dunbar, P.R.; Speiser, D.E.; Lienard, D.; Lejeune, F.; Fleischhauer, K.; Cerundolo, V.; Cerottini, J.C.; Romero, P. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 1999, 190, 705–715, doi:10.1084/jem.190.5.705.
- Brichard, V.; Van Pel, A.; Wolfel, T.; Wolfel, C.; De Plaen, E.; Lethe, B.; Coulie, P.; Boon, T. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med. 1993, 178, 489–495, doi:10.1084/jem.178.2.489.
- Kawakami, Y.; Eliyahu, S.; Delgado, C.H.; Robbins, P.F.; Sakaguchi, K.; Appella, E.; Yannelli, J.R.; Adema, G.J.; Miki, T.; Rosenberg, S.A. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA 1994, 91, 6458–6462, doi:10.1073/pnas.91.14.6458.
- Kawakami, Y.; Eliyahu, S.; Delgado, C.H.; Robbins, P.F.; Rivoltini, L.; Topalian, S.L.; Miki, T.; Rosenberg, S.A. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc. Natl. Acad. Sci. USA 1994, 91, 3515–3519, doi:10.1073/pnas.91.9.3515.
- Wang, R.F.; Appella, E.; Kawakami, Y.; Kang, X.; Rosenberg, S.A. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J. Exp. Med. 1996, 184, 2207–2216, doi:10.1084/jem.184.6.2207.
- Wang, R.F.; Robbins, P.F.; Kawakami, Y.; Kang, X.Q.; Rosenberg, S.A. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J. Exp. Med. 1995, 181, 799–804, doi:10.1084/jem.181.2.799.
- Parkhurst, M.R.; Fitzgerald, E.B.; Southwood, S.; Sette, A.; Rosenberg, S.A.; Kawakami, Y. Identification of a shared HLA-A*0201-restricted T-cell epitope from the melanoma antigen tyrosinase-related protein 2 (TRP2). Cancer Res. 1998, 58, 4895–4901.
- Olson, B.M.; Frye, T.P.; Johnson, L.E.; Fong, L.; Knutson, K.L.; Disis, M.L.; McNeel, D.G. HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase. Cancer Immunol. Immunother. 2010, 59, 943–953, doi:10.1007/s00262-010-0820-6.
- Correale, P.; Walmsley, K.; Nieroda, C.; Zaremba, S.; Zhu, M.; Schlom, J.; Tsang, K.Y. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J. Natl. Cancer Inst. 1997, 89, 293–300, doi:10.1093/jnci/89.4.293.
- Parkhurst, M.R.; Joo, J.; Riley, J.P.; Yu, Z.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin. Cancer Res. 2009, 15, 169–180, doi:10.1158/1078-0432.CCR-08-1638.
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626, doi:10.1038/mt.2010.272.
- Hammarstrom, S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999, 9, 67–81, doi:10.1006/scbi.1998.0119.
- Yee, C.; Thompson, J.A.; Roche, P.; Byrd, D.R.; Lee, P.P.; Piepkorn, M.; Kenyon, K.; Davis, M.M.; Riddell, S.R.; Greenberg, P.D. Melanocyte destruction after antigen-specific immunotherapy of melanoma: Direct evidence of t cell-mediated vitiligo. J. Exp. Med. 2000, 192, 1637–1644, doi:10.1084/jem.192.11.1637.
- Jager, E.; Maeurer, M.; Hohn, H.; Karbach, J.; Jager, D.; Zidianakis, Z.; Bakhshandeh-Bath, A.; Orth, J.; Neukirch, C.; Necker, A.; et al. Clonal expansion of Melan A-specific cytotoxic T lymphocytes in a melanoma patient responding to continued immunization with melanoma-associated peptides. Int. J. Cancer 2000, 86, 538–547, doi:10.1002/(sici)1097-0215(20000515)86:4<538::aid-ijc16>3.0.co;2-g.
- Almeida, L.G.; Sakabe, N.J.; deOliveira, A.R.; Silva, M.C.; Mundstein, A.S.; Cohen, T.; Chen, Y.T.; Chua, R.; Gurung, S.; Gnjatic, S.; et al. CTdatabase: A knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009, 37, D816-819, doi:10.1093/nar/gkn673.
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Nicolay, H.J.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182, doi:10.1016/j.molonc.2011.02.001.
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647, doi:10.1126/science.1840703.
- De Plaen, E.; Arden, K.; Traversari, C.; Gaforio, J.J.; Szikora, J.P.; De Smet, C.; Brasseur, F.; van der Bruggen, P.; Lethe, B.; Lurquin, C.; et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics 1994, 40, 360–369, doi:10.1007/bf01246677.
- Gaugler, B.; Van den Eynde, B.; van der Bruggen, P.; Romero, P.; Gaforio, J.J.; De Plaen, E.; Lethe, B.; Brasseur, F.; Boon, T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 1994, 179, 921–930, doi:10.1084/jem.179.3.921.
- Traversari, C.; van der Bruggen, P.; Luescher, I.F.; Lurquin, C.; Chomez, P.; Van Pel, A.; De Plaen, E.; Amar-Costesec, A.; Boon, T. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J. Exp. Med. 1992, 176, 1453–1457, doi:10.1084/jem.176.5.1453.
- Old, L.J.; Chen, Y.T. New paths in human cancer serology. J. Exp. Med. 1998, 187, 1163–1167, doi:10.1084/jem.187.8.1163.
- Chomez, P.; De Backer, O.; Bertrand, M.; De Plaen, E.; Boon, T.; Lucas, S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 2001, 61, 5544–5551.
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625, doi:10.1038/nrc1669.
- Lian, Y.; Meng, L.; Ding, P.; Sang, M. Epigenetic regulation of MAGE family in human cancer progression-DNA methylation, histone modification, and non-coding RNAs. Clin. Epigenetics 2018, 10, 115, doi:10.1186/s13148-018-0550-8.
- Weon, J.L.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell. Biol. 2015, 37, 1–8, doi:10.1016/j.ceb.2015.08.002.
- Van den Eynde, B.J.; van der Bruggen, P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997, 9, 684–693, doi:10.1016/s0952-7915(97)80050-7.
- Al-Khadairi, G.; Decock, J. Cancer Testis Antigens and Immunotherapy: Where Do We Stand in the Targeting of PRAME? Cancers 2019, 11, 984, doi:10.3390/cancers11070984.
- Ikeda, H.; Lethe, B.; Lehmann, F.; van Baren, N.; Baurain, J.F.; de Smet, C.; Chambost, H.; Vitale, M.; Moretta, A.; Boon, T.; et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997, 6, 199–208, doi:10.1016/s1074-7613(00)80426-4.
- Kessler, J.H.; Beekman, N.J.; Bres-Vloemans, S.A.; Verdijk, P.; van Veelen, P.A.; Kloosterman-Joosten, A.M.; Vissers, D.C.; ten Bosch, G.J.; Kester, M.G.; Sijts, A.; et al. Efficient identification of novel HLA-A(*)0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J. Exp. Med. 2001, 193, 73–88, doi:10.1084/jem.193.1.73.
- Quintarelli, C.; Dotti, G.; Hasan, S.T.; De Angelis, B.; Hoyos, V.; Errichiello, S.; Mims, M.; Luciano, L.; Shafer, J.; Leen, A.M.; et al. High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood 2011, 117, 3353–3362, doi:10.1182/blood-2010-08-300376.
- Chen, Y.T.; Scanlan, M.J.; Sahin, U.; Tureci, O.; Gure, A.O.; Tsang, S.; Williamson, B.; Stockert, E.; Pfreundschuh, M.; Old, L.J. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA 1997, 94, 1914–1918, doi:10.1073/pnas.94.5.1914.
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947, doi:10.3389/fimmu.2018.00947.
- Davis, I.D.; Chen, W.; Jackson, H.; Parente, P.; Shackleton, M.; Hopkins, W.; Chen, Q.; Dimopoulos, N.; Luke, T.; Murphy, R.; et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 10697–10702, doi:10.1073/pnas.0403572101.
- Tureci, O.; Sahin, U.; Schobert, I.; Koslowski, M.; Scmitt, H.; Schild, H.J.; Stenner, F.; Seitz, G.; Rammensee, H.G.; Pfreundschuh, M. The SSX-2 gene, which is involved in the t(X;18) translocation of synovial sarcomas, codes for the human tumor antigen HOM-MEL-40. Cancer Res. 1996, 56, 4766–4772.
- Smith, H.A.; McNeel, D.G. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin. Dev. Immunol. 2010, 2010, 150591, doi:10.1155/2010/150591.
- Boel, P.; Wildmann, C.; Sensi, M.L.; Brasseur, R.; Renauld, J.C.; Coulie, P.; Boon, T.; van der Bruggen, P. BAGE: A new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 1995, 2, 167–175, doi:10.1016/s1074-7613(95)80053-0.
- Van den Eynde, B.; Peeters, O.; De Backer, O.; Gaugler, B.; Lucas, S.; Boon, T. A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J. Exp. Med. 1995, 182, 689–698, doi:10.1084/jem.182.3.689.
- Fiszer, D.; Kurpisz, M. Major histocompatibility complex expression on human, male germ cells: A review. Am. J. Reprod. Immunol. 1998, 40, 172–176, doi:10.1111/j.1600-0897.1998.tb00409.x.
- Tyagi, P.; Mirakhur, B. MAGRIT: The largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clin. Lung Cancer 2009, 10, 371–374, doi:10.3816/CLC.2009.n.052.
- Ruiz, R.; Hunis, B.; Raez, L.E. Immunotherapeutic agents in non-small-cell lung cancer finally coming to the front lines. Curr. Oncol. Rep. 2014, 16, 400, doi:10.1007/s11912-014-0400-6.
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151, doi:10.1097/CJI.0b013e3182829903.
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61, doi:10.1016/j.cell.2014.12.033.
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74, doi:10.1126/science.aaa4971.
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200, doi:10.1146/annurev-immunol-042617-053402.
- Coulie, P.G.; Lehmann, F.; Lethe, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980, doi:10.1073/pnas.92.17.7976.
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996, 183, 1185–1192, doi:10.1084/jem.183.3.1185.
- Wolfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wolfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Buschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284, doi:10.1126/science.7652577.
- Mandruzzato, S.; Brasseur, F.; Andry, G.; Boon, T.; van der Bruggen, P. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J. Exp. Med. 1997, 186, 785–793, doi:10.1084/jem.186.5.785.
- Tashiro, H.; Brenner, M.K. Immunotherapy against cancer-related viruses. Cell Res. 2017, 27, 59–73, doi:10.1038/cr.2016.153.
- Ramos, C.A.; Narala, N.; Vyas, G.M.; Leen, A.M.; Gerdemann, U.; Sturgis, E.M.; Anderson, M.L.; Savoldo, B.; Heslop, H.E.; Brenner, M.K.; et al. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes for adoptive immunotherapy of HPV-associated malignancies. J. Immunother. 2013, 36, 66–76, doi:10.1097/CJI.0b013e318279652e.
- Carbone, A.; Gloghini, A.; Dotti, G. EBV-associated lymphoproliferative disorders: Classification and treatment. Oncologist 2008, 13, 577–585, doi:10.1634/theoncologist.2008-0036.
- Javier, R.T.; Butel, J.S. The history of tumor virology. Cancer Res. 2008, 68, 7693–7706, doi:10.1158/0008-5472.CAN-08-3301.
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding Epstein-Barr Virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022, doi:10.1158/1078-0432.CCR-14-1122-T.
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616, doi:10.1016/S2214-109X(16)30143-7.
- He, Q.; Liu, Z.; Liu, Z.; Lai, Y.; Zhou, X.; Weng, J. TCR-like antibodies in cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 99, doi:10.1186/s13045-019-0788-4.
- Bannert, N.; Hofmann, H.; Block, A.; Hohn, O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 2018, 9, 178, doi:10.3389/fmicb.2018.00178.
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219, doi:10.1038/nri.2016.27.
- Attermann, A.S.; Bjerregaard, A.M.; Saini, S.K.; Gronbaek, K.; Hadrup, S.R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 2018, 29, 2183–2191, doi:10.1093/annonc/mdy413.
- Takahashi, Y.; Harashima, N.; Kajigaya, S.; Yokoyama, H.; Cherkasova, E.; McCoy, J.P.; Hanada, K.; Mena, O.; Kurlander, R.; Tawab, A.; et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J. Clin. Investig. 2008, 118, 1099–1109, doi:10.1172/JCI34409.
- Cherkasova, E.; Scrivani, C.; Doh, S.; Weisman, Q.; Takahashi, Y.; Harashima, N.; Yokoyama, H.; Srinivasan, R.; Linehan, W.M.; Lerman, M.I.; et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. 2016, 76, 2177–2185, doi:10.1158/0008-5472.CAN-15-3139.
- Schiavetti, F.; Thonnard, J.; Colau, D.; Boon, T.; Coulie, P.G. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 2002, 62, 5510–5516.
- Rakoff-Nahoum, S.; Kuebler, P.J.; Heymann, J.J.; Sheehy, M.E.; Ortiz, G.M.; Ogg, G.S.; Barbour, J.D.; Lenz, J.; Steinfeld, A.D.; Nixon, D.F. Detection of T lymphocytes specific for human endogenous retrovirus K (HERV-K) in patients with seminoma. Aids Res. Hum. Retrovir. 2006, 22, 52–56, doi:10.1089/aid.2006.22.52.


