 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giovanni Raugei | + 5414 word(s) | 5414 | 2020-10-09 11:12:34 | | | |
| 2 | Bruce Ren | Meta information modification | 5414 | 2020-10-13 04:15:33 | | |
Video Upload Options
Despite a large number of therapeutic options available, malignant melanoma remains a highly fatal disease, especially in its metastatic forms. The oncogenic role of protein tyrosine phosphatases (PTPs) is becoming increasingly clear, paving the way for novel antitumor treatments based on their inhibition. In this review, we present the oncogenic PTPs contributing to melanoma progression and we provide, where available, a description of new inhibitory strategies designed against these enzymes and possibly useful in melanoma treatment. Considering the relevance of the immune infiltrate in supporting melanoma progression, we also focus on the role of PTPs in modulating immune cell activity, identifying interesting therapeutic options that may support the currently applied immunomodulating approaches.
1. Introduction
Reversible tyrosine phosphorylation is one of the most important post-translational modifications, which regulates key aspects of cellular biology, such as protein stability, protein–protein interactions, and enzyme activity [1] thereby modulating the functionality of fundamental elements involved in signaling transduction of mammalian cells [2]. The intracellular tyrosine phosphorylation level is maintained by a strict balance between the activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), which catalyze respectively the addition or the removal of phosphate from tyrosyl residues of their substrates [3]. An imbalance in this regulation is a well-recognized feature of several diseases, including cancer, since it induces alterations in cell growth and survival, cell migration, and tissue differentiation [4][5]. While the role of PTKs as oncogenic proteins has been largely described, allowing the development of a wide range of inhibitors already accepted in clinical use [6][7], an effective strategy to modulate PTPs for cancer therapy has yet to be identified [8].
PTPs are a large family of proteins consisting of 107 members that can be classified into four families (class I, II, III, and IV) according to the amino acid sequence at the catalytic domains [9]. Originally, PTPs were described to exclusively have a role as tumor suppressors, counteracting the activity of PTKs. However, further studies brought to light that some PTPs can also function as oncogenes depending on the availability of their functional partners and tumor type [5]. Such flexibility in functions can be explained considering that, differently from PTKs, PTPs can act both as negative and positive regulators of signal transduction pathways and can either activate or inhibit the oncogenic role of PTKs. Indeed, PTPs can exert their function by directly dephosphorylating PTKs, or, indirectly, by interfering with their downstream targets [10].
Starting from these bases, a deeper understanding of the dual role of PTPs in affecting tumor progression could lead to the development of new therapeutic strategies aimed at targeting different classes of tumors [11].
Despite the increasing evidence demonstrating the contribution of oncogenic PTPs in supporting tumor progression, the number of inhibitors available to date is still extremely limited. Primarily, this is due to the fact that PTPs have been considered for a long time as “undruggable targets”, delaying the design of pharmacological inhibitors [12][13][14]. In particular, the nature of PTP active sites represents a big challenge for the development of specific inhibitors active inside the cells [14][15]. First of all, in order to target the highly positively charged PTP active site, it would be necessary to develop negatively charged molecules, a characteristic that unfortunately strongly limits their cell permeability and bioavailability. However, promising prospects have come from the recent discovery of nonhydrolyzable, polar, and cell-permeable pTyr mimetics that gave a new chance to overcome these problems [12]. Moreover, designing specific PTP inhibitors is further complicated by the excellent conservation of the amino acid sequence inside the active site among PTPs [13]. Remarkably, although crystal structures of different members of the class-I PTPs revealed a common Cα-backbone signature, the surfaces around the PTPs’ catalytic site are characterized by distinct properties, including different topology, electrostatic potential, and lipophilic features, that can be addressed when designing novel selective inhibitors [16][17][18][19]. Furthermore, the identification of allosteric inhibitors is crucial to avoid targeting of the charged and conserved PTP active site, allowing the development of cell-permeable and selective compounds [20][21][22]. Another central obstacle is the presence of a shallow pocket at the PTPs’ catalytic site. This issue could be solved by proposing bidentate inhibitors, targeting both the active site and proximal non-conserved binding sites that are present in most of the PTPs [23][24].
Finally, another major challenge that has emerged during the screening of PTP inhibitors is the susceptibility of these enzymes to radical oxygen species (ROS), a phenomenon that in the past has often led to erroneous conclusions during investigations about possible inhibitors. Indeed, it is known that several anticancer drugs increase ROS production through redox cycling or the inhibition of ROS scavenger enzymes [25]. In analogy to other cysteine-based enzymes, the activity of PTPs is highly susceptible to oxidation of catalytic cysteine residues, which leads to the complete inactivation of these enzymes [26]. This evidence highlighted that, in order to avoid an erroneous interpretation of the mechanism of action of PTP inhibitors, their ability to generate ROS should always be evaluated both in vitro and in vivo. As far as this topic is concerned, the studies conducted in the past to analyze the mechanism of action of different types of cysteine protease inhibitors could be kept in mind as pivotal examples [27][28][29].
Noteworthy, some of the PTPs recognized to have oncogenic properties have been found to be overexpressed in highly metastatic melanoma [30][31][32][33][34][35], providing the opportunity to develop new strategies to fight this disease.
Malignant melanoma is an aggressive form of cutaneous neoplasia that derives from a series of alterations occurring in melanocytes, the melanin-producing cells resident in the basal layer of the epidermidis [36]. Melanoma retains the highest mortality rate among skin cancers and the highest potential of dissemination [37]. The majority of melanoma patients develop the cutaneous form of the disease, while non-cutaneous melanomas (which include tumors of the ocular and mucosal sites, such as anorectal, vaginal, nasal, and gastrointestinal tract) are relatively rare [38]. Both classes of melanoma are considered as a multi-factorial disease, whose pathogenesis is affected by environmental and genetic factors. However, the differential incidence of genetic alterations among melanoma subtypes and the unequal exposure to UV radiation, according to the anatomic site, strongly influence the molecular pathways involved in tumorigenesis, ultimately leading to the need for specific therapies against the different subtypes [39].
Data published in the Cancer Genome Atlas (TCGA) Network classify cutaneous melanomas into four genetic subgroups on the basis of the most frequently mutated genes involved in the mitogen-activated protein kinase (MAPK) pathway: BRAF, RAS (N-H-K), NF1, and triple wild-type (WT) melanomas [40][41]. Mutations in BRAF and NRAS are most commonly detected in primary cutaneous melanomas [42]. In particular, BRAF is mutated in about 50% of cutaneous melanomas, and among these, in 80–90% of the cases, the missense activating mutation V600E is present. Besides, NRAS mutations occur in about 20–25% of melanomas [43][44]. Following BRAF and NRAS, NF1 is the third gene most commonly mutated in cutaneous melanoma (in about 17% of the cases) and frequently co-occurs with mutations in the RASA2 gene [45][46]. NF1 mostly displays point mutations, which determine a loss of function with consequent constitutive activation of the MAPK and phosphoinositide 3-kinase (PI3K) pathways [47]. Interestingly, all these mutations finally result in a constitutive activation of MAPK/ERK signaling, which indeed is present in 98% of melanomas, promoting cellular proliferation, survival, and angiogenesis [48][49]. Finally, the loss of function of PTEN is observed in about 10–35% of cutaneous melanomas, where it confers resistance to BRAF inhibitors. This mutation results in a constitutive activation of the PI3K/AKT pathways, which, in turn, leads to cell growth and proliferation and to the inhibition of apoptosis [50].
Conversely, non-cutaneous melanomas have significantly lower numbers of mutations: Acral melanomas have, in about 15–20% of the cases, mutations in BRAF, NRAS, and KIT [51]; mucosal melanomas display KIT mutations in about 15% of the cases (primarily in genitourinary or anal forms) but rarely present mutations in BRAF and NRAS [52]; and uveal melanomas have distinct genomic patterns, presenting mutations either in GNAQ or GNA11 in > 90% of the cases while BAP1, SF3B1, and EIFAX are distinct subsets [53][54][55].
The involvement of protumoral PTPs in the oncogenic signaling pathways that characterize malignant melanoma may pave the way for new possible combination therapies based on pharmacological inhibition of oncogenic PTPs. Indeed, this approach could provide longer lasting therapeutic benefits through the inhibition of multiple nodes in the main oncogenic signaling pathways. In agreement with this hypothesis, Prahallad and co-authors found that the suppression of Src homology region 2 domain-containing phosphatase-2 (SHP-2) in BRAF mutant and in Vemurafenib-sensitive melanoma cells inhibits growth factor-induced drug resistance and delays the onset of spontaneous resistance [56]. Moreover, they identified the activating phosphorylation site on Tyr542 of SHP-2 as a valid biomarker to recognize patients with melanoma who have acquired Vemurafenib resistance due to receptor tyrosine kinases (RTKs) activation [56]. Moreover, due to the central role of SHP-2 in mutant KRAS-driven carcinogenesis, it has been demonstrated that the synergic inhibition of SHP-2 and MAPK/ERK kinase (MEK) results in decreased tumor growth in xenograft models of pancreatic ductal adenocarcinoma and non-small cell lung cancer, sustaining the utility of the dual SHP-2/MEK inhibition in KRAS mutant cancers [57].
The overall survival of patients diagnosed with advanced melanoma has strongly increased over recent years, thanks to the latest development of therapeutic strategies [58]. However, recurrence frequently occurs due to therapy failure leading to metastasis formation, representing the main cause of patient death [59]. For this reason, many efforts have been made to design new therapeutic approaches aimed at targeting the most aggressive stages of melanoma [60].
The data presented above underline that even if the PTP inhibitors available to date show only a mild effect on cell proliferation, future efforts could be made to use these compounds in combination with other pathway-targeted drugs to fight melanoma progression.
In this review, we will present a detailed overview of PTPs reported, up to date, to function as oncogenes in melanoma, either facilitating tumor progression or dampening the immune response. This information lays the foundation for the design of new therapeutic strategies specifically directed against oncogenic PTPs in melanoma [61].
2. Oncogenic Protein Tyrosine Phosphatases in Melanoma
Among the 107 known PTPs, several of them have been identified to have an oncogenic role in different types of cancers. Interestingly, recent evidence highlights their importance in supporting melanoma progression, as discussed in the following sections.
2.1. Cell Division Cycle 25 Proteins (CDC25s)
CDC25s are a family of dual-specificity phosphatases (DSPs), known to act as key regulators of cell cycle progression and DNA damage handling, through the activation of cyclin-dependent kinase (CDK) complexes. CDC25s remove inhibitory phosphates from both threonine and tyrosine residues present on the phosphate-binding loop of CDKs [62]. Their inactivation or degradation leads to cell cycle arrest [63][64]. Hence, it is not surprising that CDC25 deregulation may lead to genomic instability and cancer transformation [64]. In humans, there are three distinct CDC25 genes (CDC25A, B, and C), which specifically dephosphorylate and activate various targets [63]. All the three isoforms are involved in tumorigenesis, even if with varying extent. Specifically, the overexpression of either CDC25A and/or CDC25B has been observed in multiple tumor types, including melanoma, where it is frequently associated with a more aggressive and metastatic phenotype [65][66][67]. By contrast, the role of CDC25C seems to be less important for tumorigenesis [68][69][70]. Interestingly, Kaplan–Meier curves of melanoma patients confirm the correlation between the higher expression of all the three isoforms and a worse clinical outcome [71][72][73][74].
Considering the importance of CDC25s in facilitating cell cycle progression and cell proliferation, these proteins are potentially very interesting targets for melanoma treatment. Determination of the crystal structures of the catalytic domain of CDC25A (PDB: 1C25) [75] and CDC25B (PDB: 1QB0) [76], alongside recent progression in bioinformatic approaches, facilitated the identification of numerous compounds with inhibitory effects on these enzymes.
In particular, as far as melanoma is concerned, triptolide (TPL), a diterpene triepoxide natural compound, has been demonstrated to be active in different cancers and, specifically, on the A375.S2 melanoma cell line. Treatment with this compound induces cyclin A and CDC25A inhibition, thereby causing arrest of the cell cycle in S phase. In addition, the exposure of A375.S2 melanoma cells to TPL leads to apoptosis through caspase-8, -9, and -3 activation [77]. Moreover, cantharidin (CTD), another natural compound that shares many characteristics with TPL, shows a similar ability to inhibit cyclin A and CDC25A in the A375.S2 melanoma cell line [78].
Noteworthy, Capasso and co-workers, using both experimental and bioinformatic methods, developed several quinonoid derivatives, acting as CDC25 irreversible inhibitors . The mechanism of action of these compounds involves electrophilic modification or ROS-induced oxidation [79][80] of the catalytic cysteine residue in the PTP active site [81][82]. Among the molecules selected with this strategy, nine were identified with Ki values in the range of micro- and nano-molar and one of them (referred to as “compound 7”) showed efficacy on melanoma cells (A2058 and SAN), arresting their proliferation in G2/M phase and inducing a strong antiproliferative effect . In the same paper, the authors also assessed that treatment with compound 7 stimulates the intrinsic apoptosis pathway in a caspase-dependent manner and leads to a reduction of the CDC25C protein level (and, at a lower extent, of CDC25A). However, the mechanism of action of these quinonoid-based agents may cause many different unrelated events, due to ROS reaction with other phosphatases and with unrelated enzymes. These possibilities represent a serious limit to their therapeutic applications, due to the potential toxicity.
In order to circumvent this issue, more recently, Cerchia and co-authors performed a screening of different classes of molecules, starting from the lead inhibitor NSC28620. This approach allowed them to identify naphthylphenylketone and naphthylphenylamine derivatives, acting as CDC25 inhibitors in two aggressive human melanoma cell lines, namely A2058 and A375. In contrast with quinonoid derivatives, these compounds reversibly inhibit the enzymes (in particular, the CDC25B isoform) without generating ROS. Altogether, these characteristics make these inhibitors more interesting for possible anticancer therapy. In agreement, the reported results indicate that the treatment with these compounds affects cell cycle progression, with increased G2/M phase and reduced G0/G1 phase accumulation, by causing an increase in the phosphorylated form of cyclin-dependent kinase 1 (CDK1) [83] (Figure 1).
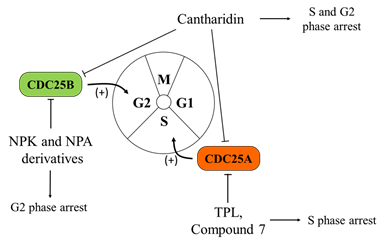
Figure 1. Effects of Cell Division Cycle 25 Proteins (CDC25s) targeting on melanoma cells. CDC25 phosphatases act as key regulators of the cell cycle, dephosphorylating cyclin-dependent kinases (CDK1, CDK2, CDK4, and CDK6) and cyclins (cyclin D, B, A, and E complexes). Several quinonoid derivatives, naphthylphenylketones and naphthylphenylamine derivatives, act as CDC25 inhibitors, arrest cells in the G0/G1 and G2/M phases of the cell cycle, and significantly inhibit the proliferation and colony formation ability of melanoma cells.
It should be underlined that the inhibitors so far cited are not closely specific for CDC25s and the question on their toxicity still remains open. Indeed, all the presented experiments, although showing clear cut results, are limited to in vitro melanoma models. More efforts are needed to confirm the efficacy of these new compounds as tools for melanoma treatment.
2.2. Low-Molecular-Weight Protein Tyrosine Phosphatase (LMW-PTP)
LMW-PTP belongs to the non-transmembrane PTPs sub-family and consists of 157 amino acids [84]. Two isoforms, generated by alternative splicing of a single gene and characterized by different activities and substrate specificities, have been found in mammalian cells [85].
Previous studies revealed that this enzyme displays a wide number of substrates, including the platelet derived growth factor (PDGF) receptor [86], insulin receptor [87], Ephrin A2 (EphA2) [88][89], and several non-receptor proteins, such as proto-oncogene tyrosine-protein kinase Src (Src) [90], focal adhesion kinase (FAK) [91], caveolin [92], signal transducer and activator of transcription 5 (STAT5) [93], β-catenin [94], and p190RhoGAP [95], thereby modulating key signaling pathways involved in tumor growth, differentiation, migration, and invasion. In this context, it is not surprising that LMW-PTP has been found to be overexpressed in several types of human [96] and rat tumors [97], where it promotes an aggressive and malignant phenotype [98][99][100].
Recently, it has been demonstrated that LMW-PTP is overexpressed in melanoma cells, contributing to the regulation of cancer cell sensitivity toward chemo- and radiotherapy. Interestingly, it has been highlighted that the treatment of melanoma cells with morin, a non-toxic natural LMW-PTP inhibitor, is able to increase the sensitivity of tumor cells toward both dacarbazine and radiotherapy. Coherently, data reported in The Human Protein Atlas database show that in melanoma patients, unfavorable prognosis is associated with high LMW-PTP expression levels, thereby confirming the role of this enzyme in regulating the in vivo survival and proliferation rate of melanoma cells [101]. Collectively, these findings suggest that LMW-PTP could be an interesting target to improve the effectiveness of anticancer treatment for melanoma patients that are naturally refractory to the therapies.
2.3. FAS-Associated Phosphatase 1 (FAP-1)
FAP-1 (or PTPN13/PTP-BAS) is a protein tyrosine phosphatase that interacts with the cytosolic portion of the Fas cell surface death receptor (FAS), whose activation leads to cell apoptosis.
FAP-1 interaction negatively regulates FAS-initiated apoptosis, preventing FAS export from the cytoplasm to the cell surface [102]. Other reported FAP-1 binding partners include the nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, α (IκBα), the Rho GTPase activated protein 1 (RhoGAP1), Ephrin B1, and the transient receptor potential cation channel M2 (TRPM2). In particular, IκBα is a putative FAP-1 substrate, being the only FAP-1-binding protein that is also dephosphorylated by this phosphatase [103].
Since FAP-1 negatively regulates FAS-initiated cell apoptosis, it has been suggested to positively affect tumor progression. Accordingly, FAP-1 has been reported to inhibit FAS-mediated apoptosis in pancreatic adenocarcinoma [104][105] and melanoma [106]. Interestingly, human melanoma cells silenced for FAP-1 show increased surface FAS expression and respond to recombinant FAS ligand (FasL) treatment by the induction of apoptosis [106]. Contrary to the possibility of blocking FAP-1 for the treatment of melanoma, there is also evidence that FAP-1 can act as a tumor suppressor in some cancer types. For example, reduced PTPN13 mRNA expression due to promoter hypermethylation or allelic loss has been observed in gastric and hepatocellular carcinomas [107][108]. Such a diversity of functions described for FAP-1, with positive and negative roles in a context-dependent manner, could be explained considering that it is among the largest intracellular PTPs, containing eight domains [109]. Despite the demonstrated role of this PTP in melanoma, to date, at least to our knowledge, no therapeutic approaches have been developed to inhibit FAP-1 in this tumor type, leaving open the possibility of designing new inhibitors.
2.4. Mitogen-Activated Protein Kinase Phosphatase-1 (MKP1)
MKP1 is a member of the threonine-tyrosine dual-specificity phosphatase family. MKP1 targets different members of the MAPK family that regulate cell proliferation and apoptosis, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK [110]. Different evidence demonstrates that MKP1-mediated JNK dephosphorylation/inactivation is essential to protect tumor cells from anticancer drug-induced apoptosis [111][112]. Interestingly, melanoma patient survival analyzed by the Kaplan–Meier curve describes a correlation between high levels of MKP1 and poor prognosis [113]. Therefore, the inhibition of MKP1 may be an effective strategy to enhance the activity of antitumor therapy [114][115]. Promising results in this context come from a study by Kundu and co-workers, who demonstrated that by combining the interferon-α2b (IFN-α2b) with a new selective MKP1 inhibitor, tyrosine phosphatase inhibitor-3 (TPI-3), it is possible to obtain better results than those achieved with IFN-α2b or TPI-3 alone in inhibiting melanoma growth both in vitro and in a xenograft nude mice model. Interestingly, the authors reported that TPI-3 is a well-tolerated compound and that mice treated with TPI-3 alone did not experience loss of weight, abnormalities in behaviors, or anatomic alterations [116] (Figure 2). All together, these findings suggest that therapeutic strategies based on the treatment with MKP1 inhibitors could contribute to improve the prognosis of patients affected by tumors expressing high MKP1 levels, such as melanoma.
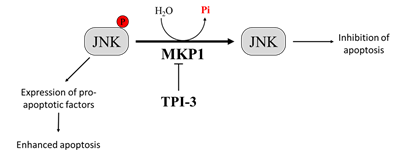
Figure 2. Effect of Mitogen-Activated Protein Kinase Phosphatase-1 (MKP1) targeting on melanoma cells. Overexpression of MKP1 phosphatase in melanoma cells contributes to enhance resistance toward anticancer drugs. For this reason, MKP1 inhibition is sufficient to enhance cancer cell death in culture and to sensitize cancer cells towards cytotoxic drugs. Among the substrates of MKP1, there is JNK. MKP1 dephosphorylates JNK, inhibiting its activation, and thereby avoiding apoptosis.
2.5. Phosphatase of Regenerating Liver (PRL)
The family of PRL consists of three members, namely PRL1, PRL2, and PRL3, and is a unique class of oncogenic dual-specificity phosphatases (alternatively known as protein tyrosine phosphatase 4A, PTP4A). Kaplan–Meier curve analysis of patients affected with melanoma shows a clear correlation between PRL phosphatase expression and poor survival [117][118].
Despite their role in cancer being well documented, the molecular functions of these proteins are still not completely understood [119]. PRL3 modulates different signaling pathways involving p53, MAPK, protein kinase B (PKB, also known as AKT), mammalian target of rapamycin (mTOR), signal transducer and activator of transcription 3 (STAT3), FAK, and vascular endothelial growth factor (VEGF), hence positively acting on tumor cell proliferation and aggressiveness [120]. PRL3 is also able to dephosphorylate the PI (4,5) P2 phosphoinositide, thereby contributing to modulation of the tumoral phenotype [121].
In addition, PRL3 promotes cell motility, invasion, and metastasis formation through different mechanisms, including its mutual activation involving the kinase Src [122], the accumulation of MMO14 matrix metalloprotease [123], and the downregulation of the tumor suppressor phosphatase and tensin homolog (PTEN), with consequent epithelial–mesenchymal transition [124]. Interestingly, in a very recent paper, PRL2 overexpression was also correlated with PTEN downregulation and poor patient survival. In particular, the authors proved that PRL2 directly downregulates PTEN by dephosphorylating its Tyr336 residue, thus increasing PTEN ubiquitination and degradation [125].
Overexpression of PRL3 has been demonstrated in many different solid tumors, including metastatic melanoma. This finding was further confirmed by Wu and colleagues, who demonstrated a higher expression of PRL3 in the metastatic melanoma cell line B16-BL6 with respect to its less metastatic counterpart B16 cells, highlighting a clear role of PRL3 in promoting metastasis formation [126]. Daouti and co-authors established that while ectopic PRL3 overexpression induces cell transformation and increases motility and invasiveness, PRL3 silencing prevents anchorage-independent cell growth in soft agar . The authors suggested that the adaptor protein p130Crk-associated substrate (p130Cas) is involved in this mechanism. Specifically, they demonstrated that treatment with thienopyridone, a selective inhibitor of all the three PRL isoforms, induces p130Cas and FAK cleavage, leading to the induction of caspase-mediated cell apoptosis and cancer cell anoikis .
Coherently, it was also shown that siRNA-mediated PRL3 depletion is able to inhibit the metastatic potential of B16-BL6 mouse melanoma cells both in vitro and in vivo [127].
Even if it has been known for a long time that a correlation exists between high PRL3 expression and metastatic risk in patients with uveal melanoma [128], only recently has a specific role for PRLs been recognized in this aggressive and metastatic tumor. In particular, collapsin response mediator protein 2 (CRMP2), a protein affecting microtubule dynamics, protein endocytosis, and vesicle recycling, has been described as a new target for PRL3 phosphatase activity. Specifically, PRL3 dephosphorylates CRMP2 on Thr514, thus enhancing cell invasiveness [129].
Considering the key role of PRL3 in mediating melanoma cell motility and metastasis formation, several attempts have been performed in order to select specific PRL3 inhibitors. Pathak and colleagues identified pentamidine [1,5-di(4-amidinophenoxy)pentane] as a relatively specific inhibitor of PRLs and tested its activity on several cancer cell lines, including the WM9 melanoma-derived cell line. Interestingly, pentamidine was also tested in nude mice, where it was able to induce marked tumor cell necrosis in engrafted WM9 human melanoma cells, without any obvious side effects [130]. In addition, the previously mentioned thienopyridone is another promising inhibitor of PRLs that has been demonstrated to be effective in reducing the aggressiveness of melanoma cells by affecting their metastatic potential (Figure 3).
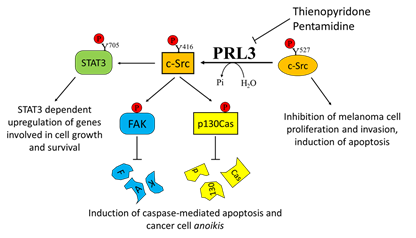
Figure 3. Effects of Phosphatase of Regenerating Liver-3 (PRL-3) targeting on melanoma progression. Elevated PRL-3 leads to Src activation through the downregulation of the synthesis of C-terminal Src kinase protein, which in turn leads to tyrosine phosphorylation of several proteins, including STAT3, FAK, and p130Cas. Thienopyridone and pentamidine derivatives, which act as PRL3 inhibitors, are effective in inhibiting melanoma cell proliferation, survival, and migration.
A possible alternative approach is based on the targeting of PRL1 trimer formation, a mechanism necessary for PRL1-mediated cell proliferation and migration [131]. Using a computer-based virtual screening, different specific compounds were selected as PRL1 trimerization inhibitors. Interestingly, one of these compounds, referred to as “Cmpd-43”, displayed a strong anticancer activity both in vitro and in vivo in a murine xenograft model of melanoma [132].
Even if further efforts are needed to improve both the effectiveness of the inhibitors described and to reduce their side effects, the reported results suggest that PRLs could be an optimal target to reduce melanoma aggressiveness.
An event that should be considered when developing new treatments targeting PRLs is its interaction with the CNNM complexes (cyclin-M family, also termed cyclin and cystathionine β-synthase (CBS) domain magnesium transport mediators). This interaction is a key node in the regulation of magnesium homeostasis [133], and cells that overexpress PRLs in complex with CNNMs accumulate intracellular magnesium [134], which favors tumor proliferation and migration [135].
A possible more promising approach has very recently been developed using a humanized antibody (PRL3-zumab) that is able to target externalized PRL3 protein on different human liver and gastric tumor cell lines, used in an orthotopic tumor model in nude mice[136]. This antibody is currently under investigation in a phase 1 clinical trial on a wide range of solid tumors and hematological malignancies (Trial Number: NCT03191682; Table.1).
Table 1. Protein tyrosine phosphatase (PTP) inhibitors involved in clinical trials for melanoma treatment.
|
Trial Number |
Compound |
Target |
Disease |
Status |
|
NCT03191682 |
PRL3-ZUMAB |
PRL3 |
Solid Tumors and Hematologic Malignancies |
Phase I |
|
NCT03114319 |
TNO155 |
SHP-2 |
Non-Small Cell Lung Cancer; Esophageal Squamous Cell Cancer (SCC); Head/Neck SCC; Melanoma |
Phase I |
|
NCT00629200 |
Sodium stibogluconate |
SHP-1 |
Malignant melanoma |
Phase I completed |
|
NCT00498979 |
Sodium stibogluconate |
SHP-1 |
Malignant melanoma |
Phase I completed |
PRL3: Phosphatase of Regenerating Liver-3; SHP-2: Src Homology Region 2 Domain-Containing Phosphatase-2; SHP-1: Src Homology Region 2 Domain-Containing Phosphatase-1
2.6. Src Homology Region 2 Domain-Containing Phosphatase-2 (SHP-2)
SHP-2, also termed tyrosine-protein phosphatase non-receptor type 11, is encoded by the PTPN11 gene [137]. SHP-2 contains two tandem SH2 domains, which act as phospho-tyrosine-binding domains and mediate the interaction of the tyrosine phosphatase with its substrates [137]. SHP-2 is auto-inhibited in the resting state, since the N-terminal SH2 domain binds to the catalytic cleft of the PTP domain, thereby blocking the access of SHP-2 substrates to the active site. Upon binding to target phospho-tyrosine residues, the N-terminal SH2 domain is released from the PTP domain and thus the enzyme is catalytically activated by reverting its auto-inhibited conformation [138].
SHP-2 is ubiquitously expressed and plays a key role in different cell signaling events, such as mitogenic activation, metabolic control, transcriptional regulation, and cell migration [139].
SHP-2 is the first tyrosine phosphatase identified as an oncogene in juvenile myelomonocytic leukemia, myelodysplastic syndromes, and acute myeloid leukemia [140]. It is remarkable that the gain-of-function mutations in SHP-2, leading to hyper-activated/deregulated mutants of the enzyme, occur in about 50% of Noonan syndrome patients [141]. Importantly, increased SHP-2 expression is a prognostic and a predictive marker of several malignancies and plays a key role in melanoma [142][143][144][145][146][147][148][149]. Indeed, this PTP has been found to be overexpressed and mutated in samples derived from melanoma patients, correlating with a strong metastatic phenotype and a poorer prognosis. These findings were further confirmed by the Kaplan–Meier curve analysis, which revealed a strong correlation between higher expression levels of SHP-2 and poor overall survival in melanoma patients [150].
Due to the involvement of SHP-2 in multiple growth factor-mediated oncogenic pathways, such as the Ras/ERK1/2 pathway, and to its fundamental role in several tumors, inhibition of SHP-2 is considered to have broad therapeutic applications in the treatment of various cancers, including melanoma [151].
The PTP inhibitor sodium stibogluconate (SSG), a drug used in the treatment of leishmaniasis [152] and identified as an inhibitor of both SHP-1 and SHP-2 [153], increases interferon-α (IFN-α)-induced signal transducer and activator of transcription 1 (STAT1) tyrosine phosphorylation and has been shown to synergize with IFN-α to inhibit WM9 human melanoma tumor growth in nude mice [154]. Accordingly, Win-Piazza and co-workers demonstrated that suppression of SHP-2 increases the antitumor activity of IFN-α2b in A375 melanoma tumor xenografts. Indeed, IFN-α2b exerts antiproliferative effects on A375 cells through STAT1/STAT2 tyrosine phosphorylation, which is negatively regulated by SHP-2. In keeping with these data, treatment with the SHP-2 inhibitor, SPI-112, increases the IFN-α2b-stimulated STAT1 phosphorylation and inhibits A375 cell growth [155].
Furthermore, Soong and colleagues revealed a peculiar role of SHP-2 in melanocytes. Specifically, Plexin B1 and tyrosine protein kinase Met (MET) assemble in an oligomeric receptor-receptor complex in melanocytes and Semaphorin-4D (Sema4D) increases this association. The consequent MET activation correlates with the transformation of melanocytes to melanoma [156]. SHP-2 mediates, at least in part, the effects downstream to the MET receptor, and this phosphatase is required for the activation of the MAPK and AKT signaling pathways in response to hepatocyte growth factor (HGF) [157][158]. The blockade of SHP-2 phosphatase activity with the inhibitor NSC-87877 reduces HGF-induced MET activation and subsequently ERK1/ERK2 and AKT phosphorylation, suggesting an important role for SHP-2 in transducing proliferative and prosurvival signals in melanocytes. Consequently, inhibition of SHP-2 can be proposed as a novel target to halt the transformation of melanocytes in melanoma.
Furthermore, SHP-2 acts as an oncogene in BRAF wild-type (either NRAS mutant or wild-type) melanoma cells. Indeed, both silencing of the activated SHP-2 E76K mutant or the administration of the allosteric SHP-2 inhibitor, SHP099, causes regression of the established melanoma, thereby suggesting that SHP-2 could be considered as a therapeutic target for BRAF wild-type melanoma.
It is widely described that HGF confers resistance to the BRAF inhibitor Vemurafenib in BRAF-mutant melanoma cells [159]. Interestingly, recent evidence underlines that SHP-2 is necessary to mediate this mechanism of resistance. Indeed, Prahallad and co-workers revealed that SHP-2 knockout clones of SK-Mel888 BRAF(V600E) mutant melanoma cells were unable to confer Vemurafenib resistance, following HGF, fibroblast growth factor 9 (FGF9), and stem cell factor (SCF) exposure. Furthermore, it has been demonstrated that SHP-2 also drives adaptive resistance to RAS viral (v-raf) oncogene homolog (RAF) and MEK inhibitors in other tumor types [160]. Accordingly, Ahmed and co-authors proved that co-targeting of MEK and SHP-2 could serve as a powerful therapeutic approach in triple-negative breast cancer and showed that SHP-2 inhibition impairs adaptive resistance to Vemurafenib in a subset of BRAF(V600E) colorectal and thyroid cancers. These results suggest that SHP-2 blockade successfully overcomes adaptive resistance to BRAF and MEK inhibitors in a defined subgroup of ERK-dependent tumors, keeping the possibility open for exploiting this strategy for melanoma treatment.
Moreover, SHP-2 acts as a scaffold protein recruiting growth factor receptor-bound protein 2/Son of Sevenless (GRB2/SOS) complex to the membrane and promoting RTK-mediated RAS activation [161][162]. It is noteworthy that the allosteric SHP-2 inhibitor SHP099 stabilizes the phosphatase in its inactive conformation, thus preventing the assembly of SHP-2 with other adaptor proteins to achieve the complete activation of RTK signaling. In keeping with this, Zhang and colleagues demonstrated that SHP-2 overexpression enhances melanoma MeWo cell viability, motility, and anchorage-independent growth, through positive regulation of the ERK1/2 and PI3K/AKT pathway . Accordingly, SHP-2 knockdown is able to revert these effects. Indeed, the specific SHP-2 inhibitor 11a-1 [163], an indole salicylic acid inhibitor, reduces the aforementioned phenomena in melanoma cells by downregulating the SHP-2-mediated ERK1/2 and AKT signaling pathways. Moreover, in vivo experiments demonstrated that 11a-1 significantly reduces xenografted melanoma tumor growth (Figure 4).
Overall, following the clear correlation between high expression levels of SHP2 and poorer survival of melanoma patients, several findings strongly suggest that SHP-2 may act as a targetable substrate against melanoma. In this perspective, SHP-2 inhibitors can be proposed as novel therapeutic approaches for melanoma treatment.
In keeping, TNO155, a recently discovered orally bioavailable SHP-2 inhibitor with antitumor activity in xenograft models [164], is currently in clinical trials for the treatment of solid tumors, including melanoma (Trial Number: NCT03114319; Table 1).
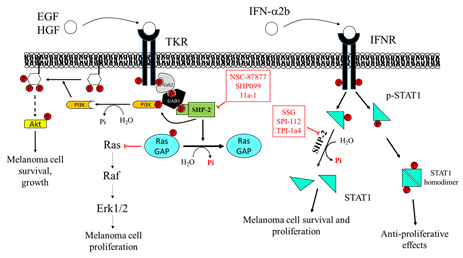
Figure 4. Effects of Src Homology Region 2 Domain-Containing Phosphatase-2 (SHP-2) targeting on melanoma cells. Left: SHP-2 has a key role in promoting the proliferation and survival of melanoma cells. SHP-2 dephosphorylates RasGAP, an inhibitor of Ras. Therefore, SHP-2, by activating Ras, promotes the RAS/RAF/ERK1/2 pathway, which sustains melanoma cell proliferation. Right: SHP-2 dephosphorylates GRB2-associated-binding protein 1 (GAB1), which releases PI3K, promoting activation of the PI3K/AKT pathway, melanoma cell growth, and survival. NSC-87877 and SHP099 inhibit SHP-2, impairing melanoma cell proliferation. SHP-2 also has an important role in regulating signaling activated by IFN-α2b. SHP-2 dephosphorylates STAT1, hindering its dimerization and its migration into the nucleus, where it stimulates the transcription of several genes, resulting in melanoma cell growth arrest. Overexpression of SHP-2 in melanoma cells blunts the response to IFN-α, favoring melanoma cell survival and dissemination. SSG, SPI-112, and TPI-1a4 are potent inhibitors of SHP-2, enhancing the anti-proliferative activity of IFN-α.
References
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146, doi:10.1016/j.ceb.2009.01.028.
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L. Lo The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280, doi:10.3892/ijmm.2017.3036.
- Östman, A. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001, 11, 258–266, doi:10.1016/S0962-8924(01)01990-0.
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365, doi:10.1038/35077225.
- Motiwala, T.; Jacob, S.T. Role of Protein Tyrosine Phosphatases in Cancer. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 297–329.
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731, doi:10.3390/cancers12030731.
- Angelucci, A. Targeting Tyrosine Kinases in Cancer: Lessons for an Effective Targeted Therapy in the Clinic. Cancers 2019, 11, 490, doi:10.3390/cancers11040490.
- Kim, M.; Baek, M.; Kim, D.J. Protein Tyrosine Signaling and its Potential Therapeutic Implications in Carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226–4246, doi:10.2174/1381612823666170616082125.
- Alonso, A.; Nunes-Xavier, C.E.; Bayón, Y.; Pulido, R. The extended family of protein tyrosine phosphatases. In Protein Tyrosine Phosphatases; Humana Press: New York, NY, USA, 2016; pp. 1–23.
- Julien, S.G.; Dubé, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49, doi:10.1038/nrc2980.
- Ventura, J.-J.; Nebreda, Á.R. Protein kinases and phosphatases as therapeutic targets in cancer. Clin. Transl. Oncol. 2006, 8, 153–160, doi:10.1007/s12094-006-0005-0.
- Zhang, Z.-Y. Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc. Chem. Res. 2017, 50, 122–129, doi:10.1021/acs.accounts.6b00537.
- Scott, L.M.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr. Pharm. Des. 2010, 16, 1843–1862, doi:10.2174/138161210791209027.
- Stanford, S.M.; Bottini, N. Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci. 2017, 38, 524–540, doi:10.1016/j.tips.2017.03.004.
- Barr, A.J. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2010, 2, 1563–1576, doi:10.4155/fmc.10.241.
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711, doi:10.1016/j.cell.2004.05.018.
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Møller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136, doi:10.1128/MCB.21.21.7117-7136.2001.
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.F.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Müller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363, doi:10.1016/j.cell.2008.11.038.
- Lawrence, H.R.; Pireddu, R.; Chen, L.; Luo, Y.; Sung, S.-S.; Szymanski, A.M.; Yip, M.L.R.; Guida, W.C.; Sebti, S.M.; Wu, J.; et al. Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J. Med. Chem. 2008, 51, 4948–4956, doi:10.1021/jm8002526.
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737, doi:10.1038/nsmb803.
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566, doi:10.1038/nchembio.1528.
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152, doi:10.1038/nature18621.
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425, doi:10.1073/pnas.94.25.13420.
- Sun, J.-P.; Fedorov, A.A.; Lee, S.-Y.; Guo, X.-L.; Shen, K.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.-Y. Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J. Biol. Chem. 2003, 278, 12406–12414, doi:10.1074/jbc.M212491200.
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591, doi:10.1038/nrd2803.
- Ostman, A.; Frijhoff, J.; Sandin, A.; Böhmer, F.-D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356, doi:10.1093/jb/mvr104.
- Ohayon, S.; Refua, M.; Hendler, A.; Aharoni, A.; Brik, A. Harnessing the oxidation susceptibility of deubiquitinases for inhibition with small molecules. Angew. Chem. Int. Ed. Engl. 2015, 54, 599–603, doi:10.1002/anie.201408411.
- Gopinath, P.; Mahammed, A.; Ohayon, S.; Gross, Z.; Brik, A. Understanding and predicting the potency of ROS-based enzyme inhibitors, exemplified by naphthoquinones and ubiquitin specific protease-2. Chem. Sci. 2016, 7, 7079–7086, doi:10.1039/c6sc02758j.
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva-Jr, F.P. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207, doi:10.1186/s12935-019-0925-8.
- Daouti, S.; Li, W.; Qian, H.; Huang, K.-S.; Holmgren, J.; Levin, W.; Reik, L.; McGady, D.L.; Gillespie, P.; Perrotta, A.; et al. A Selective Phosphatase of Regenerating Liver Phosphatase Inhibitor Suppresses Tumor Cell Anchorage-Independent Growth by a Novel Mechanism Involving p130Cas Cleavage. Cancer Res. 2008, 68, 1162–1169, doi:10.1158/0008-5472.CAN-07-2349.
- Lori, G.; Paoli, P.; Caselli, A.; Cirri, P.; Marzocchini, R.; Mangoni, M.; Talamonti, C.; Livi, L.; Raugei, G. Targeting LMW-PTP to sensitize melanoma cancer cells toward chemo- and radiotherapy. Cancer Med. 2018, 7, 1933–1943, doi:10.1002/cam4.1435.
- Tang, L.; Li, G.; Tron, V.A.; Trotter, M.J.; Ho, V.C. Expression of cell cycle regulators in human cutaneous malignant melanoma. Melanoma Res. 1999, 9, 148, doi:10.1097/00008390-199904000-00006.
- Cheng, Y.-P.; Chiu, H.-Y.; Hsiao, T.-L.; Hsiao, C.-H.; Lin, C.-C.; Liao, Y.-H. Scalp melanoma in a woman with LEOPARD syndrome: Possible implication of PTPN11 signaling in melanoma pathogenesis. J. Am. Acad. Dermatol. 2013, 69, e186–e187, doi:10.1016/j.jaad.2013.04.033.
- Hill, K.S.; Roberts, E.R.; Wang, X.; Marin, E.; Park, T.D.; Son, S.; Ren, Y.; Fang, B.; Yoder, S.; Kim, S.; et al. PTPN11 Plays Oncogenic Roles and Is a Therapeutic Target for BRAF Wild-Type Melanomas. Mol. Cancer Res. 2019, 17, 583–593, doi:10.1158/1541-7786.MCR-18-0777.
- Zhang, R.-Y.; Yu, Z.-H.; Zeng, L.; Zhang, S.; Bai, Y.; Miao, J.; Chen, L.; Xie, J.; Zhang, Z.-Y. SHP2 phosphatase as a novel therapeutic target for melanoma treatment. Oncotarget 2016, 7, 73817–73829, doi:10.18632/oncotarget.12074.
- Jackett, L.A.; Scolyer, R.A. A Review of Key Biological and Molecular Events Underpinning Transformation of Melanocytes to Primary and Metastatic Melanoma. Cancers 2019, 11, 2041, doi:10.3390/cancers11122041.
- Hartman, R.I.; Lin, J.Y. Cutaneous Melanoma-A Review in Detection, Staging, and Management. Hematol. Oncol. Clin. North. Am. 2019, 33, 25–38, doi:10.1016/j.hoc.2018.09.005.
- McLaughlin, C.C.; Wu, X.-C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of noncutaneous melanomas in the U.S. Cancer 2005, 103, 1000–1007, doi:10.1002/cncr.20866.
- Wilkins, D.K.; Nathan, P.D. Therapeutic opportunities in noncutaneous melanoma. Ther. Adv. Med. Oncol. 2009, 1, 29–36, doi:10.1177/1758834009337664.
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; Baboud, J. Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696, doi:10.1016/j.cell.2015.05.044.
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180, doi:10.1038/nature22071.
- Rajkumar, S.; Watson, I.R. Molecular characterisation of cutaneous melanoma: Creating a framework for targeted and immune therapies. Br. J. Cancer 2016, 115, 145–155, doi:10.1038/bjc.2016.195.
- Ribas, A.; Flaherty, K.T. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat. Rev. Clin. Oncol. 2011, 8, 426–433, doi:10.1038/nrclinonc.2011.69.
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867, doi:10.1016/s0092-8674(04)00215-6.
- Peltonen, S.; Kallionpää, R.A.; Peltonen, J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp. Dermatol. 2017, 26, 645–648, doi:10.1111/exd.13212.
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002, doi:10.1038/ng.3361.
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157, doi:10.1038/labinvest.2016.142.
- Chiappetta, C.; Proietti, I.; Soccodato, V.; Puggioni, C.; Zaralli, R.; Pacini, L.; Porta, N.; Skroza, N.; Petrozza, V.; Potenza, C.; et al. BRAF and NRAS mutations are heterogeneous and not mutually exclusive in nodular melanoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2015, 23, 172–177, doi:10.1097/PAI.0000000000000071.
- Griffin, M.; Scotto, D.; Josephs, D.H.; Mele, S.; Crescioli, S.; Bax, H.J.; Pellizzari, G.; Wynne, M.D.; Nakamura, M.; Hoffmann, R.M.; et al. BRAF inhibitors: Resistance and the promise of combination treatments for melanoma. Oncotarget 2017, 8, 78174–78192, doi:10.18632/oncotarget.19836.
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760, doi:10.1158/0008-5472.CAN-10-2954.
- Yeh, I.; Jorgenson, E.; Shen, L.; Xu, M.; North, J.P.; Shain, A.H.; Reuss, D.; Wu, H.; Robinson, W.A.; Olshen, A.; et al. Targeted Genomic Profiling of Acral Melanoma. J. Natl. Cancer Inst. 2019, 111, 1068–1077, doi:10.1093/jnci/djz005.
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346, doi:10.1200/JCO.2006.06.2984.
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. New Engl. J. Med. 2010, 363, 2191–2199, doi:10.1056/NEJMoa1000584.
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234, doi:10.1167/iovs.08-2145.
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat. Commun. 2018, 9, 116, doi:10.1038/s41467-017-02428-w.
- Prahallad, A.; Heynen, G.J.; Germano, G.; Willems, S.M.; Evers, B.; Vecchione, L.; Gambino, V.; Lieftink, C.; Beijersbergen, R.L.; Di Nicolantonio, F.; et al. PTPN11 is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell Rep. 2015, 12, 1978–1985, doi:10.1016/j.celrep.2015.08.037.
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960, doi:10.1038/s41591-018-0024-8.
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482, doi:10.1038/nrclinonc.2017.43.
- Damsky, W.E.; Theodosakis, N.; Bosenberg, M. Melanoma metastasis: New concepts and evolving paradigms. Oncogene 2014, 33, 2413–2422, doi:10.1038/onc.2013.194.
- Mouawad, R.; Sebert, M.; Michels, J.; Bloch, J.; Spano, J.-P.; Khayat, D. Treatment for metastatic malignant melanoma: Old drugs and new strategies. Crit. Rev. Oncol. Hematol. 2010, 74, 27–39, doi:10.1016/j.critrevonc.2009.08.005.
- Jiang, Z.-X.; Zhang, Z.-Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272, doi:10.1007/s10555-008-9113-3.
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507, doi:10.1038/nrc2169.
- Capasso, A.; Cerchia, C.; Di Giovanni, C.; Granato, G.; Albano, F.; Romano, S.; De Vendittis, E.; Ruocco, M.R.; Lavecchia, A. Ligand-based chemoinformatic discovery of a novel small molecule inhibitor targeting CDC25 dual specificity phosphatases and displaying in vitro efficacy against melanoma cells. Oncotarget 2015, 6, 40202–40222, doi:10.18632/oncotarget.5473.
- Karlsson-Rosenthal, C.; Millar, J.B.A. Cdc25: Mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006, 16, 285–292, doi:10.1016/j.tcb.2006.04.002.
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.-Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z.; et al. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126, doi:10.1016/j.celrep.2018.03.039.
- Cangi, M.G.; Cukor, B.; Soung, P.; Signoretti, S.; Moreira, G.; Ranashinge, M.; Cady, B.; Pagano, M.; Loda, M. Role of the Cdc25A phosphatase in human breast cancer. J. Clin. Investig. 2000, 106, 753–761, doi:10.1172/JCI9174.
- Ma, Z.-Q.; Chua, S.S.; DeMayo, F.J.; Tsai, S.Y. Induction of mammary gland hyperplasia in transgenic mice over-expressing human Cdc25B. Oncogene 1999, 18, 4564–4576, doi:10.1038/sj.onc.1202809.
- Kristjánsdóttir, K.; Rudolph, J. Cdc25 Phosphatases and Cancer. Chem. Biol. 2004, 11, 1043–1051, doi:10.1016/j.chembiol.2004.07.007.
- Albert, H.; Santos, S.; Battaglia, E.; Brito, M.; Monteiro, C.; Bagrel, D. Differential expression of CDC25 phosphatases splice variants in human breast cancer cells. Clin. Chem. Lab. Med. 2011, 49, doi:10.1515/CCLM.2011.635.
- Bahassi, E.M.; Hennigan, R.F.; Myer, D.L.; Stambrook, P.J. Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation. Oncogene 2004, 23, 2658–2663, doi:10.1038/sj.onc.1207425.
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, doi:10.1126/science.aan2507.
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000164045-CDC25A/pathology/melanoma (accessed on 27 August 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000101224-CDC25B/pathology/melanoma (accessed on 27 August 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000158402-CDC25C/pathology/melanoma (accessed on 27 August 2020).
- Fauman, E.B.; Cogswell, J.P.; Lovejoy, B.; Rocque, W.J.; Holmes, W.; Montana, V.G.; Piwnica-Worms, H.; Rink, M.J.; Saper, M.A. Crystal Structure of the Catalytic Domain of the Human Cell Cycle Control Phosphatase, Cdc25A. Cell 1998, 93, 617–625, doi:10.1016/S0092-8674(00)81190-3.
- Reynolds, R.A.; Yem, A.W.; Wolfe, C.L.; Deibel, M.R.; Chidester, C.G.; Watenpaugh, K.D. Crystal structure of the catalytic subunit of Cdc25B required for G 2 /M phase transition of the cell cycle 1 1Edited by I. A. Wilson. J. Mol. Biol. 1999, 293, 559–568, doi:10.1006/jmbi.1999.3168.
- Hung, F.-M.; Chen, Y.-L.; Huang, A.-C.; Hsiao, Y.-P.; Yang, J.-S.; Chung, M.-T.; Chueh, F.-S.; Lu, H.-F.; Chung, J.-G. Triptolide induces S phase arrest via the inhibition of cyclin E and CDC25A and triggers apoptosis via caspase- and mitochondrial-dependent signaling pathways in A375.S2 human melanoma cells. Oncol. Rep. 2013, 29, 1053–1060, doi:10.3892/or.2013.2230.
- Hsiao, Y.-P.; Tsai, C.-H.; Wu, P.-P.; Hsu, S.-C.; Liu, H.-C.; Huang, Y.-P.; Yang, J.-H.; Chung, J.-G. Cantharidin induces G2/M phase arrest by inhibition of Cdc25c and Cyclin A and triggers apoptosis through reactive oxygen species and the mitochondria-dependent pathways of A375.S2 human melanoma cells. Int. J. Oncol. 2014, 45, 2393–2402, doi:10.3892/ijo.2014.2689.
- Kar, S.; Lefterov, I.M.; Wang, M.; Lazo, J.S.; Scott, C.N.; Wilcox, C.S.; Carr, B.I. Binding and Inhibition of Cdc25 Phosphatases by Vitamin K Analogues †. Biochemistry 2003, 42, 10490–10497, doi:10.1021/bi027418p.
- Pu, L.; Amoscato, A.A.; Bier, M.E.; Lazo, J.S. Dual G 1 and G 2 Phase Inhibition by a Novel, Selective Cdc25 Inhibitor 7-Chloro-6-(2-morpholin-4-ylethylamino)- quinoline-5,8-dione. J. Biol. Chem. 2002, 277, 46877–46885, doi:10.1074/jbc.M207902200.
- Brisson, M.; Nguyen, T.; Wipf, P.; Joo, B.; Day, B.W.; Skoko, J.S.; Schreiber, E.M.; Foster, C.; Bansal, P.; Lazo, J.S. Redox Regulation of Cdc25B by Cell-Active Quinolinediones. Mol. Pharmacol. 2005, 68, 1810–1820, doi:10.1124/mol.105.016360.
- Zhou, Y.; Feng, X.; Wang, L.; Du, J.; Zhou, Y.; Yu, H.; Zang, Y.; Li, J.; Li, J. LGH00031, a novel ortho-quinonoid inhibitor of cell division cycle 25B, inhibits human cancer cells via ROS generation. Acta Pharmacol. Sin. 2009, 30, 1359–1368, doi:10.1038/aps.2009.131.
- Cerchia, C.; Nasso, R.; Mori, M.; Villa, S.; Gelain, A.; Capasso, A.; Aliotta, F.; Simonetti, M.; Rullo, R.; Masullo, M.; et al. Discovery of Novel Naphthylphenylketone and Naphthylphenylamine Derivatives as Cell Division Cycle 25B (CDC25B) Phosphatase Inhibitors: Design, Synthesis, Inhibition Mechanism, and in Vitro Efficacy against Melanoma Cell Lines. J. Med. Chem. 2019, 62, 7089–7110, doi:10.1021/acs.jmedchem.9b00632.
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846, doi:10.1038/nrm2039.
- Caselli, A.; Paoli, P.; Santi, A.; Mugnaioni, C.; Toti, A.; Camici, G.; Cirri, P. Low molecular weight protein tyrosine phosphatase: Multifaceted functions of an evolutionarily conserved enzyme. Biochim. Biophys. Acta 2016, 1864, 1339–1355, doi:10.1016/j.bbapap.2016.07.001.
- Chiarugi, P.; Cirri, P.; Raugei, G.; Manao, G.; Taddei, L.; Ramponi, G. Low M(r) phosphotyrosine protein phosphatase interacts with the PDGF receptor directly via its catalytic site. Biochem. Biophys. Res. Commun. 1996, 219, 21–25, doi:10.1006/bbrc.1996.0174.
- Chiarugi, P.; Cirri, P.; Marra, F.; Raugei, G.; Camici, G.; Manao, G.; Ramponi, G. LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling. Biochem. Biophys. Res. Commun. 1997, 238, 676–682, doi:10.1006/bbrc.1997.7355.
- Kikawa, K.D.; Vidale, D.R.; Van Etten, R.L.; Kinch, M.S. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J. Biol. Chem. 2002, 277, 39274–39279, doi:10.1074/jbc.M207127200.
- Chiarugi, P.; Taddei, M.L.; Schiavone, N.; Papucci, L.; Giannoni, E.; Fiaschi, T.; Capaccioli, S.; Raugei, G.; Ramponi, G. LMW-PTP is a positive regulator of tumor onset and growth. Oncogene 2004, 23, 3905–3914, doi:10.1038/sj.onc.1207508.
- Zambuzzi, W.F.; Granjeiro, J.M.; Parikh, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Ferreira, C.V. Modulation of Src activity by low molecular weight protein tyrosine phosphatase during osteoblast differentiation. Cell. Physiol. Biochem. 2008, 22, 497–506, doi:10.1159/000185506.
- Rigacci, S.; Rovida, E.; Dello Sbarba, P.; Berti, A. Low Mr phosphotyrosine protein phosphatase associates and dephosphorylates p125 focal adhesion kinase, interfering with cell motility and spreading. J. Biol. Chem. 2002, 277, 41631–41636, doi:10.1074/jbc.M201709200.
- Caselli, A.; Taddei, M.L.; Bini, C.; Paoli, P.; Camici, G.; Manao, G.; Cirri, P.; Ramponi, G. Low molecular weight protein tyrosine phosphatase and caveolin-1: Interaction and isoenzyme-dependent regulation. Biochemistry 2007, 46, 6383–6392, doi:10.1021/bi0620858.
- Rigacci, S.; Talini, D.; Berti, A. LMW-PTP associates and dephosphorylates STAT5 interacting with its C-terminal domain. Biochem. Biophys. Res. Commun. 2003, 312, 360–366, doi:10.1016/j.bbrc.2003.10.126.
- Taddei, M.L.; Chiarugi, P.; Cirri, P.; Buricchi, F.; Fiaschi, T.; Giannoni, E.; Talini, D.; Cozzi, G.; Formigli, L.; Raugei, G.; et al. Β-Catenin Interacts With Low-Molecular-Weight Protein Tyrosine Phosphatase Leading To Cadherin-Mediated Cell-Cell Adhesion Increase. Cancer Res. 2002, 62, 6489–6499.
- Chiarugi, P.; Cirri, P.; Taddei, L.; Giannoni, E.; Camici, G.; Manao, G.; Raugei, G.; Ramponi, G. The low M(r) protein-tyrosine phosphatase is involved in Rho-mediated cytoskeleton rearrangement after integrin and platelet-derived growth factor stimulation. J. Biol. Chem. 2000, 275, 4640–4646, doi:10.1074/jbc.275.7.4640.
- Malentacchi, F.; Marzocchini, R.; Gelmini, S.; Orlando, C.; Serio, M.; Ramponi, G.; Raugei, G. Up-regulated expression of low molecular weight protein tyrosine phosphatases in different human cancers. Biochem. Biophys. Res. Commun. 2005, 334, 875–883, doi:10.1016/j.bbrc.2005.06.176.
- Marzocchini, R.; Malentacchi, F.; Biagini, M.; Cirelli, D.; Luceri, C.; Caderni, G.; Raugei, G. The expression of low molecular weight protein tyrosine phosphatase is up-regulated in 1,2-dimethylhydrazine-induced colon tumours in rats. Int. J. Cancer 2008, 122, 1675–1678, doi:10.1002/ijc.23266.
- Ferreira, P.A.; Ruela-de-Sousa, R.R.; Queiroz, K.C.S.; Souza, A.C.S.; Milani, R.; Pilli, R.A.; Peppelenbosch, M.P.; den Hertog, J.; Ferreira, C.V. Knocking down low molecular weight protein tyrosine phosphatase (LMW-PTP) reverts chemoresistance through inactivation of Src and Bcr-Abl proteins. PLoS ONE 2012, 7, e44312, doi:10.1371/journal.pone.0044312.
- Capitani, N.; Lori, G.; Paoli, P.; Patrussi, L.; Troilo, A.; Baldari, C.T.; Raugei, G.; D’Elios, M.M. LMW-PTP targeting potentiates the effects of drugs used in chronic lymphocytic leukemia therapy. Cancer Cell Int. 2019, 19, 67, doi:10.1186/s12935-019-0786-1.
- Alho, I.; Costa, L.; Bicho, M.; Coelho, C. Low molecular weight protein tyrosine phosphatase isoforms regulate breast cancer cells migration through a RhoA dependent mechanism. PLoS ONE 2013, 8, e76307, doi:10.1371/journal.pone.0076307.
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000143727-ACP1/pathology/melanoma (accessed on 27 August 2020).
- Sato, T.; Irie, S.; Kitada, S.; Reed, J. FAP-1: A protein tyrosine phosphatase that associates with Fas. Science 1995, 268, 411–415, doi:10.1126/science.7536343.
- Nakai, Y.; Irie, S.; Sato, T.-A. Identification of IκBα as a substrate of Fas-associated phosphatase-1. Eur. J. Biochem. 2000, 267, 7170–7175, doi:10.1046/j.1432-1327.2000.01818.x.
- Ungefroren, H.; Voss, M.; Jansen, M.; Roeder, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Human pancreatic adenocarcinomas express Fas and Fas ligand yet are resistant to Fas-mediated apoptosis. Cancer Res. 1998, 58, 1741–1749.
- Ungefroren, H.; Kruse, M.L.; Trauzold, A.; Roeschmann, S.; Roeder, C.; Arlt, A.; Henne-Bruns, D.; Kalthoff, H. FAP-1 in pancreatic cancer cells: Functional and mechanistic studies on its inhibitory role in CD95-mediated apoptosis. J. Cell Sci. 2001, 114, 2735–2746.
- Ivanov, V.N.; Lopez Bergami, P.; Maulit, G.; Sato, T.-A.; Sassoon, D.; Ronai, Z. FAP-1 association with Fas (Apo-1) inhibits Fas expression on the cell surface. Mol. Cell. Biol. 2003, 23, 3623–3635, doi:10.1128/mcb.23.10.3623-3635.2003.
- Ying, J.; Li, H.; Cui, Y.; Wong, A.H.Y.; Langford, C.; Tao, Q. Epigenetic disruption of two proapoptotic genes MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas through hypermethylation of a common bidirectional promoter. Leukemia 2006, 20, 1173–1175, doi:10.1038/sj.leu.2404193.
- Yeh, S.-H.; Wu, D.-C.; Tsai, C.-Y.; Kuo, T.-J.; Yu, W.-C.; Chang, Y.-S.; Chen, C.-L.; Chang, C.-F.; Chen, D.-S.; Chen, P.-J. Genetic characterization of fas-associated phosphatase-1 as a putative tumor suppressor gene on chromosome 4q21.3 in hepatocellular carcinoma. Clin. Cancer Res. 2006, 12, 1097–1108, doi:10.1158/1078-0432.CCR-05-1383.
- Abaan, O.D.; Toretsky, J.A. PTPL1: A large phosphatase with a split personality. Cancer Metastasis Rev. 2008, 27, 205–214, doi:10.1007/s10555-008-9114-2.
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in tumorigenesis, tumor progression and therapy. Cancer Med. 2016, 5, 2061–2068, doi:10.1002/cam4.772.
- Chattopadhyay, S.; Machado-Pinilla, R.; Manguan-García, C.; Belda-Iniesta, C.; Moratilla, C.; Cejas, P.; Fresno-Vara, J.A.; de Castro-Carpeño, J.; Casado, E.; Nistal, M.; et al. MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene 2006, 25, 3335–3345, doi:10.1038/sj.onc.1209364.
- Wang, Z.; Xu, J.; Zhou, J.-Y.; Liu, Y.; Wu, G.S. Mitogen-Activated Protein Kinase Phosphatase-1 Is Required for Cisplatin Resistance. Cancer Res. 2006, 66, 8870–8877, doi:10.1158/0008-5472.CAN-06-1280.
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000120129-DUSP1/pathology/melanoma (accessed on 27 August 2020).
- Liao, Q.; Guo, J.; Kleeff, J.; Zimmermann, A.; Büchler, M.W.; Korc, M.; Friess, H. Down-regulation of the dual-specificity phosphatase MKP-1 suppresses tumorigenicity of pancreatic cancer cells. Gastroenterology 2003, 124, 1830–1845, doi:10.1016/S0016-5085(03)00398-6.
- Mizuno, R.; Oya, M.; Shiomi, T.; Marumo, K.; Okada, Y.; Murai, M. Inhibition of MKP-1 expression potentiates JNK related apoptosis in renal cancer cells. J. Urol. 2004, 172, 723–727, doi:10.1097/01.ju.0000124990.37563.00.
- Kundu, S.; Fan, K.; Cao, M.; Lindner, D.J.; Tuthill, R.; Liu, L.; Gerson, S.; Borden, E.; Yi, T. Tyrosine phosphatase inhibitor-3 sensitizes melanoma and colon cancer to biotherapeutics and chemotherapeutics. Mol. Cancer Ther. 2010, 9, 2287–2296, doi:10.1158/1535-7163.MCT-10-0159.
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000184007-PTP4A2/pathology/melanoma (accessed on 27 August 2020).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000184489-PTP4A3/pathology/melanoma (accessed on 27 August 2020).
- Wei, M.; Korotkov, K.V.; Blackburn, J.S. Targeting phosphatases of regenerating liver (PRLs) in cancer. Pharmacol. Ther. 2018, 190, 128–138, doi:10.1016/j.pharmthera.2018.05.014.
- Duciel, L.; Monraz Gomez, L.C.; Kondratova, M.; Kuperstein, I.; Saule, S. The Phosphatase PRL-3 Is Involved in Key Steps of Cancer Metastasis. J. Mol. Biol. 2019, 431, 3056–3067, doi:10.1016/j.jmb.2019.06.008.
- McParland, V.; Varsano, G.; Li, X.; Thornton, J.; Baby, J.; Aravind, A.; Meyer, C.; Pavic, K.; Rios, P.; Köhn, M. The metastasis-promoting phosphatase PRL-3 shows activity toward phosphoinositides. Biochemistry 2011, 50, 7579–7590, doi:10.1021/bi201095z.
- Fiordalisi, J.J.; Dewar, B.J.; Graves, L.M.; Madigan, J.P.; Cox, A.D. Src-Mediated Phosphorylation of the Tyrosine Phosphatase PRL-3 Is Required for PRL-3 Promotion of Rho Activation, Motility and Invasion. PLoS ONE 2013, 8, e64309, doi:10.1371/journal.pone.0064309.
- Maacha, S.; Anezo, O.; Foy, M.; Liot, G.; Mery, L.; Laurent, C.; Sastre-Garau, X.; Piperno-Neumann, S.; Cassoux, N.; Planque, N.; et al. Protein Tyrosine Phosphatase 4A3 (PTP4A3) Promotes Human Uveal Melanoma Aggressiveness Through Membrane Accumulation of Matrix Metalloproteinase 14 (MMP14). Investig. Opthalmology Vis. Sci. 2016, 57, 1982, doi:10.1167/iovs.15-18780.
- Wang, H.; Quah, S.Y.; Dong, J.M.; Manser, E.; Tang, J.P.; Zeng, Q. PRL-3 Down-regulates PTEN Expression and Signals through PI3K to Promote Epithelial-Mesenchymal Transition. Cancer Res. 2007, 67, 2922–2926, doi:10.1158/0008-5472.CAN-06-3598.
- Li, Q.; Bai, Y.; Lyle, L.T.; Yu, G.; Amarasinghe, O.; Nguele Meke, F.; Carlock, C.; Zhang, Z.-Y. Mechanism of PRL2 phosphatase-mediated PTEN degradation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 20538–20548, doi:10.1073/pnas.2002964117.
- Wu, X.; Zeng, H.; Zhang, X.; Zhao, Y.; Sha, H.; Ge, X.; Zhang, M.; Gao, X.; Xu, Q. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am. J. Pathol. 2004, 164, 2039–2054, doi:10.1016/S0002-9440(10)63763-7.
- Qian, F.; Li, Y.-P.; Sheng, X.; Zhang, Z.-C.; Song, R.; Dong, W.; Cao, S.-X.; Hua, Z.-C.; Xu, Q. PRL-3 siRNA inhibits the metastasis of B16-BL6 mouse melanoma cells in vitro and in vivo. Mol. Med. 2007, 13, 151–159, doi:10.2119/2006–00076
- Laurent, C.; Valet, F.; Planque, N.; Silveri, L.; Maacha, S.; Anezo, O.; Hupe, P.; Plancher, C.; Reyes, C.; Albaud, B.; et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011, 71, 666–674, doi:10.1158/0008-5472.CAN-10-0605.
- Duciel, L.; Anezo, O.; Mandal, K.; Laurent, C.; Planque, N.; Coquelle, F.M.; Gentien, D.; Manneville, J.-B.; Saule, S. Protein tyrosine phosphatase 4A3 (PTP4A3/PRL-3) promotes the aggressiveness of human uveal melanoma through dephosphorylation of CRMP2. Sci. Rep. 2019, 9, 2990, doi:10.1038/s41598-019-39643-y.
- Pathak, M.K.; Dhawan, D.; Lindner, D.J.; Borden, E.C.; Farver, C.; Yi, T. Pentamidine is an inhibitor of PRL phosphatases with anticancer activity. Mol. Cancer Ther. 2002, 1, 1255–1264.
- Sun, J.-P.; Luo, Y.; Yu, X.; Wang, W.-Q.; Zhou, B.; Liang, F.; Zhang, Z.-Y. Phosphatase activity, trimerization, and the C-terminal polybasic region are all required for PRL1-mediated cell growth and migration. J. Biol. Chem. 2007, 282, 29043–29051, doi:10.1074/jbc.M703537200.
- Bai, Y.; Yu, Z.H.; Liu, S.; Zhang, L.; Zhang, R.Y.; Zeng, L.F.; Zhang, S.; Zhang, Z.Y. Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res. 2016, 76, 4805–4815, doi:10.1158/0008-5472.CAN-15-2323.
- Giménez-Mascarell, P.; González-Recio, I.; Fernández-Rodríguez, C.; Oyenarte, I.; Müller, D.; Martínez-Chantar, M.; Martínez-Cruz, L. Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators. Int. J. Mol. Sci. 2019, 20, 1135, doi:10.3390/ijms20051135.
- Hardy, S.; Kostantin, E.; Hatzihristidis, T.; Zolotarov, Y.; Uetani, N.; Tremblay, M.L. Physiological and oncogenic roles of the PRL phosphatases. FEBS J. 2018, 285, 3886–3908, doi:10.1111/febs.14503.
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbé, D.P.; Bégin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 2015, 34, 986–995, doi:10.1038/onc.2014.33.
- Thura, M.; Al-Aidaroos, A.Q.; Gupta, A.; Chee, C.E.; Lee, S.C.; Hui, K.M.; Li, J.; Guan, Y.K.; Yong, W.P.; So, J.; et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat. Commun. 2019, 10, 2484, doi:10.1038/s41467-019-10127-x.
- Freeman, R.M.; Plutzky, J.; Neel, B.G. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: A putative homolog of Drosophila corkscrew. Proc. Natl. Acad. Sci. USA, 1992, 89, 11239–11243, doi:10.1073/pnas.89.23.11239.
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal Structure of the Tyrosine Phosphatase SHP-2. Cell 1998, 92, 441–450, doi:10.1016/S0092-8674(00)80938-1.
- QU, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288, doi:10.1038/sj.cr.7290055.
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150, doi:10.1038/ng1156.
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468, doi:10.1038/ng772.
- Hu, Z.-Q.; Ma, R.; Zhang, C.; Li, J.; Li, L.; Hu, Z.-T.; Gao, Q.; Li, W.-M. Expression and clinical significance of tyrosine phosphatase SHP2 in thyroid carcinoma. Oncol. Lett. 2015, 10, 1507–1512, doi:10.3892/ol.2015.3479.
- Dong, S.; Li, F.-Q.; Zhang, Q.; Lv, K.-Z.; Yang, H.-L.; Gao, Y.; Yu, J.-R. Expression and Clinical Significance of SHP2 in Gastric Cancer. J. Int. Med. Res. 2012, 40, 2083–2089, doi:10.1177/030006051204000605.
- Leibowitz, M.S.; Srivastava, R.M.; Andrade Filho, P.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 Is Overexpressed and Inhibits pSTAT1-Mediated APM Component Expression, T-cell Attracting Chemokine Secretion, and CTL Recognition in Head and Neck Cancer Cells. Clin. Cancer Res. 2013, 19, 798–808, doi:10.1158/1078-0432.CCR-12-1517.
- Hu, Z.; Fang, H.; Wang, X.; Chen, D.; Chen, Z.; Wang, S. Overexpression of SHP2 tyrosine phosphatase promotes the tumorigenesis of breast carcinoma. Oncol. Rep. 2014, 32, 205–212, doi:10.3892/or.2014.3201.
- Xie, H.; Huang, S.; Li, W.; Zhao, H.; Zhang, T.; Zhang, D. Upregulation of Src homology phosphotyrosyl phosphatase 2 (Shp2) expression in oral cancer and knockdown of Shp2 expression inhibit tumor cell viability and invasion in vitro. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 234–242, doi:10.1016/j.oooo.2013.10.018.
- Han, T.; Xiang, D.-M.; Sun, W.; Liu, N.; Sun, H.-L.; Wen, W.; Shen, W.-F.; Wang, R.-Y.; Chen, C.; Wang, X.; et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J. Hepatol. 2015, 63, 651–660, doi:10.1016/j.jhep.2015.03.036.
- Zheng, J.; Huang, S.; Huang, Y.; Song, L.; Yin, Y.; Kong, W.; Chen, X.; Ouyang, X. Expression and prognosis value of SHP2 in patients with pancreatic ductal adenocarcinoma. Tumor Biol. 2016, 37, 7853–7859, doi:10.1007/s13277-015-4675-5.
- Zhang, K.; Zhao, H.; Ji, Z.; Zhang, C.; Zhou, P.; Wang, L.; Chen, Q.; Wang, J.; Zhang, P.; Chen, Z.; et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene 2016, 35, 1271–1282, doi:10.1038/onc.2015.184.
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000179295-PTPN11/pathology/melanoma (accessed on 27 August 2020).
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.-Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, doi:10.1021/acs.jmedchem.0c00249.
- Berman, J.D. Human leishmaniasis: Clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin. Infect. Dis. 1997, 24, 684–703, doi:10.1093/clind/24.4.684.
- Pathak, M.K.; Yi, T. Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J. Immunol. 2001, 167, 3391–3397, doi:10.4049/jimmunol.167.6.3391.
- Yi, T.; Pathak, M.K.; Lindner, D.J.; Ketterer, M.E.; Farver, C.; Borden, E.C. Anticancer Activity of Sodium Stibogluconate in Synergy with IFNs. J. Immunol. 2002, 169, 5978–5985, doi:10.4049/jimmunol.169.10.5978.
- Win-Piazza, H.; Schneeberger, V.E.; Chen, L.; Pernazza, D.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Enhanced anti-melanoma efficacy of interferon alfa-2b via inhibition of Shp2. Cancer Lett. 2012, 320, 81–85, doi:10.1016/j.canlet.2012.01.034.
- Soong, J.; Scott, G. Plexin B1 inhibits MET through direct association and regulates Shp2 expression in melanocytes. J. Cell Sci. 2013, 126, 688–695, doi:10.1242/jcs.119487.
- Li, J.; Reed, S.A.; Johnson, S.E. Hepatocyte growth factor (HGF) signals through SHP2 to regulate primary mouse myoblast proliferation. Exp. Cell Res. 2009, 315, 2284–2292, doi:10.1016/j.yexcr.2009.04.011.
- Schaeper, U.; Gehring, N.H.; Fuchs, K.P.; Sachs, M.; Kempkes, B.; Birchmeier, W. Coupling of Gab1 to C-Met, Grb2, and Shp2 Mediates Biological Responses. J. Cell Biol. 2000, 149, 1419–1432, doi:10.1083/jcb.149.7.1419.
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509, doi:10.1038/nature11249.
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019, 26, 65–78, doi:10.1016/j.celrep.2018.12.013.
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 2008, 20, 453–459, doi:10.1016/j.cellsig.2007.10.002.
- Grossmann, K.S.; Rosário, M.; Birchmeier, C.; Birchmeier, W. The Tyrosine Phosphatase Shp2 in Development and Cancer. Adv. Cancer Res. 2010, 106, 53–89.
- Zeng, L.-F.; Zhang, R.-Y.; Yu, Z.-H.; Li, S.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Mali, R.S.; Li, X.; Chan, R.J.; et al. Therapeutic Potential of Targeting the Oncogenic SHP2 Phosphatase. J. Med. Chem. 2014, 57, 6594–6609, doi:10.1021/jm5006176.
- LaMarche, M.J.; Acker, M.G.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.; Chen, Y.-N.P.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, doi:10.1021/acs.jmedchem.0c01170.


