 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mirko Minini | + 3671 word(s) | 3671 | 2020-10-05 16:06:16 | | | |
| 2 | Vivi Li | -25 word(s) | 3646 | 2020-10-10 08:19:43 | | | | |
| 3 | Vivi Li | Meta information modification | 3646 | 2020-10-14 08:32:14 | | |
Video Upload Options
Inositol and its phosphate metabolites play a pivotal role in several biochemical pathways and gene expression regulation: inositol pyrophosphates (PP-IPs) have been increasingly appreciated as key signaling modulators. Fluctuations in their intracellular levels hugely impact the transfer of phosphates and the phosphorylation status of several target proteins. Pharmacological modulation of the proteins associated with PP-IP activities has proved to be beneficial in various pathological settings. IP7 has been extensively studied and found to play a key role in pathways associated with PP-IP activities. Three inositol hexakisphosphate kinase (IP6K) isoforms regulate IP7 synthesis in mammals. Genomic deletion or enzymic inhibition of IP6K1 has been shown to reduce cell invasiveness and migration capacity, protecting against chemical-induced carcinogenesis. IP6K1 could therefore be a useful target in anticancer treatment.
1. Introduction
Inositol is a ubiquitous polyol involved in a number of essential processes in living organisms. Myo-inositol is physiologically the most important of nine isomers and is the precursor of a bewildering number of complex inositol-containing molecules, including inositol phosphates [1][2]. Inositol compounds are essential for many biological functions in living cells: membrane biogenesis [3], trafficking [4], signal transduction, and regulation of gene expression [5]. Inositol phosphates are prominent mediators of these processes. Inositol-1,4,5-trisphosphate (IP3) has been widely investigated as an intracellular second messenger [6][7][8]. It is metabolized to a large number of additional inositol polyphosphates that also function as cell signals [9]. Among these, inositol hexakisphosphate (IP6), also known as phytic acid, is the most abundant inositol polyphosphate found in eukaryotes, identified as the principal phosphate-storage molecule in plant seeds [10][11]. It is involved in regulation of trafficking [12] as well as in several nuclear events [13][14]. Inositol hexakisphosphate is the building block to which successive phosphate groups are added to yield inositol pyrophosphates (PP-IPs) [15][16], where as many as one or two energetic di(β)phosphates bonds are crammed around the six-carbon inositol ring [17]. This class of molecule recently gained appreciation as critical modulators of a huge number of “signaling” pathways [18][19]. As proof of concept, PP-IPs show high turnover as their intracellular levels fluctuate significantly in various pathological disorders, including cancer [20].
2. IP6Ks: Balance, Activity, and Regulation in Physiological Homeostasis and Cancer
IP6Ks have been identified in several organisms [21][22][23]. In mammals, the three isoforms identified [24][25] have distinct sequences that are selectively involved in protein–protein interactions and post-translational modifications [25]. These regions of IP6Ks protein sequence regulate the activity, stability, subcellular distribution, and target proteins of IP6Ks [26][27]. The isoforms also differ in tissue expression. In humans, IP6K1 is widely expressed, while IP6K2 is higher in the breast, thymus, colon, adipose tissue, testis, prostate, and smooth muscle. In heart and skeletal muscle, IP6K3 is the most expressed form [28]. The IP6Ks belong to the same family of inositol phosphate kinases as IP3K (IP3-kinase) and IPMK (inositol phosphate multikinase), all characterized by a common PxxxDxKxG motif in the inositol binding region [29]. On the contrary, PPIP5K1 and PPIP5K2—homologs of the yeast enzyme Vip1—do not belong to the inositol phosphate kinase family, as they have a histidine acid phosphatase-like domain in the C-terminal portion of the protein in addition to the kinase domain [30].
IP6Ks can phosphorylate IP6 to 5-IP7 and IP5 to PP-IP4 [31]. It is arguable that the relative affinities of a given IP6K for IP6 over IP5 vary in different organisms, from yeast to mammals. For instance, in humans, IP6K2 displays a 20-fold higher affinity for IP6 than for IP5, while IP6K1 shows a 5-fold higher KM (concentration of substrates when the reaction reaches half of Vmax) for IP6 than for IP5 [21].
Furthermore, measurement of IP6Ks has advantages with respect to direct quantitation of PP-IPs. Estimation of inositol PP-IPs suffers from a number of problems, including intrinsically higher chemical reactivity and a higher degradation rate, which can be ascribed to the intrinsic acidic phosphatase domain of PPIP5K and to the hydrolytic activity exerted by DIPP (diphosphoinositol-phosphate phosphohydrolase) proteins [32]. Indeed, previous studies have been unable to detect a change in PP-IPs in response to biochemical/metabolic stimuli [17], although further investigations have provided compelling evidence in support of this hypothesis [33]. On the other hand, noncatalytic functions of IP6K could make tricky the association with PP-IPs signaling. It has also been demonstrated that PP-IPs turn over rapidly (recruiting up to 50% of the IP6 pool), depending on chemical (ATP and fluoride) stimulus [16] or during specific cell phase transitions, such as those of the cell cycle [34].
The activity of IP6K is closely coupled to activation of G protein signaling. G protein-coupled receptor (GPCR) activation through overexpression of Gαq fosters phospholipase-C-dependent release of IP3 by phosphatidyl-inositol-bisphosphate (PIP2) cleavage [35]. In turn, the increased availability of IP3 provides the substrate for inositol kinases to produce a plethora of inositol phosphates (chiefly, IP6 and IP5) and inositol pyrophosphates (PP-IPs). Overexpression of IP6K only results in a minimal increase in PP-IPs, even in the presence of high levels of IP5 and IP6, while when IP6K is overexpressed together with GPCR activation, a significantly increased release of PP-IPs has been recorded [35]. These findings suggest a cooperative network linking GPCR and IP6Ks, which can tune inositol metabolism by acting as an “IPK-dependent IP code” [35]. This hypothesis has contributed to a revision of the role traditionally attributed to IP6. It is widely agreed that inositol hexakisphosphate displays a bewildering number of physiological and pharmacological activities [10]. However, the IPK-dependent IP code hypothesis may substantiate the suggestion made 20 years ago by Shears [12] who proposed that the critical importance of IP6 may depend on being a tipping point between IP3 and the successive generation of IPs. Indeed, increasing evidence in recent years has provided sound confirmation that it is the further phosphorylation of IP6 to IPs that yields physiologically active metabolites [36]. Any factor that potentiates IP3 release through phospholipase-C activation is likely to reduce PIP2 levels while promoting inositol phosphokinase (IP6K) activity. Accordingly, phospholipase-C and IP6K both seem to play a potentially critical role in several biological pathways.
2.1. IP6K1
IP6K1 has been implicated in biological processes, such as energy metabolism, insulin signaling, trafficking, chromatin remodeling, cell migration, cancer metastasis, and neutrophil functions.
Recent studies suggest that in IP6K1-KO mice models, IP6K1 suppression increases energy expenditure by stimulating the protein kinase AMPK [37][38]. AMPK and Akt are significantly modulated under insulin stimulation [39]. IP6K1 could modulate AMPK and Akt activities by interfering with insulin release. The link between IP6K1 and Akt merits detailed discussion. Akt resides in the cytosol in an inactive conformation and translocases to the plasma membrane after cell stimulation. The Akt pleckestrin homology domain has a high affinity for PIP3, which promotes Akt translocation to the membrane [40]. The Akt/PI3K interaction causes conformational changes and subsequent PDK1-dependent phosphorylation at the Thr308 kinase domain. However, full activation requires a further phosphorylation at S473, catalyzed by several enzymes, including PDK2 and ILK. IP7 competitively binds to the PH domain, thus preventing its phosphorylation and activation by PDK1. Notably, IP7 strongly inhibits Akt activation, with an IC50 of 20 nM, close to the Kd (35 nM) displayed by PIP3 in respect to the PH domain of Akt [41]. IP6K1 knockout leads to increased PDK1-dependent Akt activation, determining a plethora of biochemical consequences for metabolic regulation, not yet well investigated. Indeed, after glucose stimulation and subsequent increase in the ATP/ADP ratio, a significant increase in IP7 was observed. In detail, IP7 production by IP6K1 inhibits the stimulatory effect of IP6 on AMPK. The response of IP7 to the increase in ATP/ADP ratio occurs a few minutes (10–30) after the stimulus. In turn, IP7 associates with the Akt PH domain, preventing interaction with PIP3 and therefore reducing Akt membrane translocation and consequent insulin-stimulated glucose uptake. This mechanism involves feedback, whereby increased availability of ATP drives the system to inhibit glucose uptake by modulating insulin transduction by blocking Akt membrane recruitment [42][43][44]. This regulation may also be indirectly affected by IP7-promoted nuclear localization of LKB1. Nuclear transfer of LKB reduces LKB cytosolic activity, thus hindering AMPK phosphorylation and activation [45]. It is worth noting that RNAi silencing of IP6K1 blocks IP7 and insulin release after glucose stimulation. In IP6K1-KO models, changes in the intracellular IP6/IP7 ratio increase AMPK activation [46]. Conversely, Akt signaling is significantly increased, leading to a decrease in GSK3b phosphorylation, and augmented protein translation. Reduction in GSK3b phosphorylation increases its catalytic activity and is likely be followed by a surge in adipogenesis and diminished glycogen levels [47]. Indeed, after insulin stimulation, IP7 decreases (from 33% to 60%) in IP6K1 knockout hepatocytes, whereas Akt and GSK3β increase, improving glucose tolerance, presumably due to a decrease in hepatic glucose production [48]. Conversely, overexpression of IP6K1 finally impairs insulin-signaling transduction, whereas IP6K1 silencing may lead to insulin hypersensitivity, as observed in IP6K1 KO mice. As proof of concept, a number of animal models of insulin hypersensitivity share the common biochemical signature of an increased tier of Akt activation and translocation [49]. Furthermore, in mouse embryo fibroblasts (MEFs), IP6K1-induced energy expenditure inhibition leads to reduction of glycolysis via IP7-mediated destabilization of the interaction between the transcriptional activators of glycolytic genes (GCR1 and GCR2) [50].
Although IP6K2 proves sensitive to ATP/ADP fluctuations and may induce IP7 synthesis, it is unlikely that it could act as a sensor of energy requirements, as does IP6K. This apparent conundrum can be explained if we consider the cell compartmentalization of IP6K. In fact, while IP6K1 is usually found in the cytosol and nucleus, IP6K2 is almost all in the nucleus [51].
2.2. IP6K2
A number of studies suggest an essential role for IP6K2 in cell death, migration, cancer metastasis, and progression. IP6K2 activity sensitizes a number of cancer cells, including OVCAR3, HeLa, HEK293, PC12, and HL60, to apoptosis [52][53][54][55]. Deletion of IP6K2 prevents apoptotic consequences of γ-irradiation or β-interferon addition to ovarian cancer cells, while overexpression of IP6K2 significantly raises cell death rate under the same conditions [53]. Overexpression of IP6K2 augments the cytotoxic effects of many cell stressors, whereas transfection with a dominant negative IP6K2 decreases cell death. It is noteworthy that the apoptosis surge is associated with increased synthesis of IP7 and transfer of IP6K2 from nuclei to mitochondria, while no changes are recorded in the intracellular localization of the other IP6K isoforms [52]. In detail, IP6K2 directly mediates IFNβ-induced apoptosis [52] by enzymically regulating p53 activity and by increasing expression of the Apo2L/TRAIL ligand that initiates apoptosis through death-receptor signaling. Namely, HSP90 physiologically binds IP6K2 and inhibits its catalytic activity. By interfering with HSP-IP6K2 binding, HSP90 fosters IP6K2 activation that ultimately leads to increased cell apoptosis [56]. Nuclear localization of IP6K2, promoted by interaction with HSP90, is a mandatory step for establishing proper IP6K2-p53 binding. [57]. Indeed, IP6K2 has been demonstrated to directly modulate p53-dependent apoptosis. Gene disruption of IP6K2 in colorectal cancer cells selectively impairs p53-mediated cell death and favors cell cycle arrest [57]. This interaction suppresses phosphorylation of the cell cycle arrest regulator (p21) and its transcription, while enhancing p53-mediated apoptosis [58]. This implies that IP6K2 acts as a switching factor, driving p53 activity towards apoptosis rather than cell cycle arrest. It should be noted that although IP6K2 regulates p53 by direct binding, its catalytic activity generating IP7 is essential for its influence on p53 signaling. It has also been observed that IP6K2 can promote apoptosis independently of its enzyme activity. By interacting with TRAF2, IP6K2 interferes with apoptosis and nuclear factor kappa β (NF-kβ) signaling, thus affecting the release of tumor necrosis factor α (TNFα) [27]. The proapoptotic activity of IP6K2 is successfully antagonized by heat-shock proteins (HSPs). Overall, these findings suggest that IP6K2 actively participates in the regulation of the Apo2L/TRAIL cell death pathway. Moreover, PP-IPs modulate cell death and telomere length in yeast by antagonizing the homolog of ataxia telangiectasia mutated (ATM) kinase, a regulator of the DNA damage response and apoptosis in mammals [59].
As strong as IP6K2-mediated apoptosis may be, IP6K2 participation in the regulation of such functions through its nuclear [60], mitochondrial [53], and cytosolic [54][61] localization requires further investigation.
As observed in IP6K1-KO models, IP6K2-KO, too, reduces cell–cell adhesion, growth, spreading, metastasis, and FAK phosphorylation in cancer cells. The molecular mechanisms so far proposed include LKB1 sequestering in the nucleus and inhibition of cytosolic phosphatase activation, and consequently, FAK dephosphorylation [59]. Remarkably, IP6K1 and IP6K2 both favor sequestering of LKB into the nucleus in an inactive form [45][61].
The tumor suppressor LKB1 is credited with inhibiting FAK activation [62] and enhancing E-cadherin expression [63], thus inhibiting motility and invasiveness. These findings strongly suggest that LKB1 plays a critical role in controlling the balance between cell–cell and cell–matrix adhesion. In addition, by modulating AMPK activity, LKB1 interferes with a number of critical metabolic processes [64]. Interaction with two subunits of the heterotrimeric holoenzyme (STRAD and Mo25) in the cytosol leads to phosphorylation of LKB1 at serine-428 and then activation by PKCδ [65]. This finding is worth mentioning as it suggests that IP6K2/IP7 can fine tune the activity of “constitutive” kinases, like PKCδ and CK2 [37], as previously indicated.
Indeed, a number of results have clearly established that deletion of IP6K1 or IP6K2 reduces cell migration, while IP6K2-KO, quite paradoxically, reduces tumor volume [66]. IP6K2-KO cells display almost total loss of IP8 levels, whereas only a small decrease in IP8 levels was recorded in IP6K1-KO [58][67]. It is tempting to speculate that persistent IP7 synthesis, even at a lower rate, is mandatory for apoptosis, as previously suggested. However, somewhat paradoxically, complete suppression of IP6K2 enhances development of carcinoma of the gastrointestinal tract in mice [68], probably because IP6K2-dependent pyrophosphate synthesis may in turn activate p53 and protein kinase CK2, thus promoting apoptosis [2]. In IP6K2 knockout mice, a substantial increase in tumorigenesis in response to 4-nitroquinoline-1-oxide, a UV-mimetic carcinogen, has been observed [69]. These findings provide indirect confirmation of the link between IP6K2 and p53, as p53-mediated apoptosis is required for apoptosis induced by UV-mimetic factors. However, unlike p53 knockouts models, the IP6K2 mutants do not develop spontaneous tumors. This apparently odd behavior suggests that IP6K2 may only influence p53 proapoptotic activity when the system is exposed to a carcinogen stressor but does not directly entail “spontaneous” carcinogenesis.
In ovarian carcinoma cells, IP6K2 deletion confers protection against interferon alpha (IFNα)-induced cell death, whereas overexpression of IP6K2 enhances the apoptosis rate promoted by IFNα and/or γ-irradiation [70]. Yet some controversial results have also been reported, since under estradiol stimulation, β-catenin-induced oncogenesis significantly increases IP6K2 gene expression downstream of the Wnt/β-catenin signaling pathway [71]. Overexpression of IP6K2 presumably leads to increased pyrophosphate synthesis, reducing cell levels of IP6, which may in turn contribute to the transformed phenotype. On the other hand, suppression of IP6K1 confers protection against tumors experimentally induced with carcinogens [72].
Although these findings are still preliminary, they suggest that IP6K1 and IP6K2 can exert opposite effects in carcinogenesis. It is also likely that the effects of IP6K2 on cancer cells are disjointed, i.e., IP6K2 probably enhances apoptosis while increasing the acquisition of an invading/migrating phenotype. IP6K2 may, therefore, act as a tumor suppressor in the initiation stage but contribute to metastatic spread by enacting EMT at later stages. It is worth underlining that similar dual roles have been observed for TGF-β1 [73].
2.3. IP6K3
IP6K3 is highly expressed in mouse and human myotubes and muscle [74]. Its physiological role is relatively unexplored. High levels of expression have been detected in the brain. Purkinje cells regulate motor learning and coordination, and IP6K3 deletion alters these functions. Abnormalities in cell size and spine density are detected, perturbed by dysfunctional IP6K3 binding of adducin and spectrin, two cytoskeletal proteins involved in the morphogenesis of dendritic trees [74]. Regarding other IP6Ks, IP6K3 seems to participate somehow in glucose metabolism. Indeed, IP6K3-null mice exhibit lower blood glucose and reduced insulin levels, associated with increased plasma lactate levels. These findings suggest that downregulation or suppression of IP6K3 can enhance glycolysis. However, IP6K3 suppression is followed by a significant reduction in pyruvate dehydrogenase kinase-4 (PDK4) [75]. Since PDK4 depresses glucose oxidation by inhibiting conversion of pyruvate to acetyl coenzyme A (acetyl-CoA), it is paradoxical that IP6K3 suppression does not lead to an increase in glucose oxidation.
3. Future Perspectives
Growing interest focused on IPs has shed light on their biological functions and corresponding deregulation issues. Among IPs, IP7 plays a significant role in cell metabolic balance, ATP production, and phosphate homeostasis. From these studies, IP6Ks emerge as key regulators of IP7 intracellular levels in physiological and pathological processes (Figure 2).
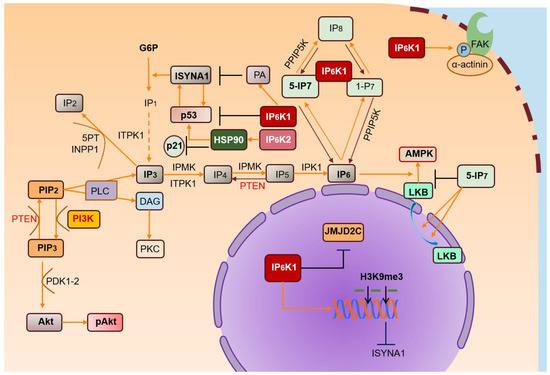
Figure 2. IP6Ks and their pathways. IP3 is metabolized in many inositol polyphosphates, of which IP6 is the most abundant. IP6Ks produce IPs (IP7) starting from IP6. IP6K/IP7 levels are crucial for regulating various biological processes. IP6K1 binds α-actinin localized at focal adhesions, promoting its phosphorylation by FAK and regulating cell migration. IP6-stimulated AMPK activation is inhibited by high levels of IP7, reducing cytosolic localization of LKB. High PA levels promote nuclear IP6K1 translocation, inhibiting ISYNA1, and consequently, de novo biosynthesis of myo-inositol. Nuclear IP6K1 interacts with JMJD2C and induces its dissociation from chromatin, increasing H3K9me3 levels and inhibiting transcription of target genes. Likewise, IP6K2 may localize in the nucleus, downstream of its interaction with HSP90. In turn, nuclear IP6K2 localization promotes binding to p53, suppressing p21 activation and transcription.
Interest in the development of molecular factors that can (selectively) interrogate and manipulate the cell actions of inositol pyrophosphates, especially by modulating IP6Ks and PPIP5Ks, is gaining momentum [70]. Targeting these pathways could be helpful in certain diseases but also potentially dangerous. For example, knockout experiments on IP6Ks highlighted a worse situation in mice, sensitizing the animals to chemical tumorigenesis [69], lung inflammation [76], and loss of motor learning, coordination, and fitness [74][77]. It is therefore crucial to determine whether pharmacological inhibition of IP6Ks is safe enough to pursue clinical investigations.
Studies based on gene deletion assays are unlikely to provide useful data, since more than 900 genes are altered by deletion of IP6K homolog (Kcs1) in S. cerevisiae [78]. The range of this genetic penetration probably highlights the functional polyvalence of IP6Ks, which presumably have both catalytic and scaffolding functions, as already demonstrated for inositol pentakisphosphate kinase [79] and inositol polyphosphate multikinase [80]. A more promising approach may focus on specific cell-permeant inhibitors of PP-IPs or on “physiological” modulators of IP6Ks, an approach that at least in principle would not be flawed by secondary genetic changes or interference with IP6K scaffolding functions.
The compound N2-(m-trifluorobenzyl)N6-(p-nitrobenzyl)purine (TNP) has been shown to bind specifically to IP6Ks by competing with ATP for the same binding site. As a result, TNP reduces IP7 levels by inhibiting the kinase and phosphatase activities of IP6Ks. Within 2 h of treating various cell types with 10–30 μM TNP, levels of IP7 fell by 60–90% [81][82], and IP8 synthesis was also significantly reduced [83]. As expected, IP6 levels increased proportionally by as much as 40%.
TNP does not efficiently cross the blood–brain and blood–testis barriers. In fact, chronic TNP administration (15 weeks, 10 mg/kg/day) in mice does not lead to neuronal or reproductive abnormalities [84]. However, TNP could interfere with the metabolism of other drugs by inducing modifications in drug signaling or increasing Ca2+ and Zn2+ levels [85].
TNP inhibitory activity discriminates between IP6Ks and other inositol phosphate kinases (IPMKs and IP3Ks). The catalytic site of the IP6K family is structurally related to that of IPMKs and IP3Ks, though IP6Ks have around 100-fold lower affinity for ATP than do the latter [82]. Higher TNP values are therefore required to efficiently neutralize IP3K (IC50 0.47 μM for IP6Ks versus 18 μM for IP3K). However, TNP displays some off-target effects, including ERK phosphorylation, which in principle is not mediated by IP6Ks. The use of TNP to investigate the intracellular functions of IP6Ks is therefore debatable. To minimize undesirable effects, it could be useful to develop safe and selective inhibitors of IP6K isoforms for investigating the specific role sustained by the different IP6K isoforms.
Regarding carcinogenesis, IP6K1 and IP6K2 activities presumably drive cells and tissues towards opposite outcomes. As previously reported, IP6K1 joins in Akt signaling, and its knockout decreases IP7 synthesis, resulting in enhanced PDK-dependent phosphorylation of Akt activation. Hyperactivation of Akt (∼10- to 50-fold) [86][87] is known to enable tumorigenesis [88]. However, IP6K1-KO is only associated with a minimal increase in Akt activation in mice [37], insufficient to enact neoplastic development [37]. Indeed, it has been reported that deletion of IP6K1 protects against chemical tumorigenesis and metastasis [67], although the mechanisms underlying the effect are still unknown. Instead, IP6K2-KO sensitizes to chemical tumorigenesis and probably increases the occurrence of spontaneous cancer [72].
Acronyms: 1-IP7 (1-diphospho-2,3,4,5,6-pentakisphosphate); 5-IP7 (5-diphospho-1,2,3,4,6-pentakisphosphate); Akt (protein kinase B); AMPK (5’ AMP-activated protein kinase); DAG (diacylglycerol); FAK (focal adhesion kinase); H3K9me3 (histone 3 lysine 9 trimethylation); IP2 (inositol-2-phosphate); IP3 (inositol-3-phosphate); IP4 (inositol-4-phosphate); IP5 (inositol-5-phosphate); IP6 (inositol-hexakisphosphate or phytic acid); IP6K1 and IP6K2 (inositol hexakisphosphate kinase 1/2); IPK1 (inositol-pentakisphosphate 2-kinase); IPMK (inositol polyphosphate multikinase); ISYNA1 (d-3-myoinositol-phosphate synthase); JMJD2C (Jumonji domain-containing protein 2C); LKB (liver kinase B1); P (phosphate group); PA (phosphatidic acid); PI3K (phosphatidylinositol 3-kinase); PIP2 (phosphatidyl-inositol-4,5-biphosphate); PIP3 (phosphatidylinositol-3-phosphate); PKC (protein kinase C); PLC (phospholipase C); PPIP5K (inositol hexakisphosphate and diphosphoinositol-pentakisphosphate kinase); PTEN (phosphatase and tensin homolog); P8 (1,5-bis-diphosphoinositol 2,3,4,6-tetrakisphosphate); G6P (glucose-6-phosphate); IP1 (inositol-1-phosphate, myo-Inositol); ITPK1 (inositol-tetrakisphosphate 1 kinase).
References
- Thota, S.G.; Bhandari, R. The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. J. Biosci. 2015, 40, 593–605.
- Bizzarri, M.; Fuso, A.; Dinicola, S.; Cucina, A.; Bevilacqua, A. Pharmacodynamics and pharmacokinetics of inositol(s) in health and disease. Expert Opin. Drug Metab. Toxicol. 2016, 14, 1–16.
- Roth, M.G. Phosphoinositides in constitutive membrane traffic. Physiol. Rev. 2004, 84, 699–730.
- Martin, T.F. Phosphoinositide lipids as signaling molecules: Common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu. Rev. Cell Dev. Biol. 1998, 14, 231–264.
- Monserrate, J.P.; York, J.D. Inositol phosphate synthesis and the nuclear processes they affect. Curr. Opin. Cell Biol. 2010, 22, 365–373.
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21.
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The calcium entry pas de deux. Science 2000, 287, 1604–1605.
- Irvine, R.F. 20 years of Ins(1,4,5)P3, and 40 years before. Nat. Rev. Mol. Cell Biol. 2003, 4, 586–590.
- Michell, R.H. Inositol derivatives: Evolution and functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 151–161.
- Raboy, V. Myo-Inositol-1,2,3,4,5,6-hexakisphosphate. Phytochemistry 2003, 64, 1033–1043.
- Plimmer, R.H.; Page, H.J. An investigation of Phytin. Biochem. J. 1913, 7, 157–174.
- Shears, S.B. Assessing the omnipotence of inositol hexakisphosphate. Cell Signal. 2001, 13, 151–158.
- Hanakahi, L.A.; Bartlet-Jones, M.; Chappell, C.; Pappin, D.; West, S.C. Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell 2000, 102, 721–729.
- Macbeth, M.R.; Schubert, H.L.; Vandemark, A.P.; Lingam, A.T.; Hill, C.P.; Bass, B.L. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 2005, 309, 1534–1539.
- Stephens, L.; Radenberg, T.; Thiel, U.; Vogel, G.; Khoo, K.H.; Dell, A.; Jackson, T.R.; Hawkins, P.T.; Mayr, G.W. The detection, purification, structural characterization and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s). J. Biol. Chem. 1993, 268, 4009–4015.
- Menniti, F.S.; Miller, R.N.; Putney, J.W., Jr.; Shears, S.B. Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. J. Biol. Chem. 1993, 268, 3850–3856.
- Shears, S.B. Inositol pyrophosphates: Why so many phosphates? Adv. Biol. Regul. 2015, 57, 203–216.
- Wilson, M.; Livermore, T.; Saiardi, A. Inositol pyrophosphates: Between signalling and metabolism. Biochem. J. 2013, 452, 369–379.
- Shears, S.B. Diphosphoinositol polyphosphates: Metabolic messengers? Mol. Pharmacol. 2009, 76, 236–252.
- Glennon, M.C.; Shears, S.B. Turnover of inositol pentakisphosphates, inositol hexakisphosphate and diphosphoinositol polyphosphates in primary cultured hepatocytes. Biochem. J. 1993, 293, 583–590.
- Saiardi, A.; Erdjument-Bromage, H.; Snowman, A.M.; Tempst, P.; Snyder, S.H. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 1999, 9, 1323–1326.
- Saiardi, A.; Caffrey, J.J.; Snyder, S.H.; Shears, S.B. The inositol hexakisphosphate kinase family: Catalytic flexibility and function in yeast vacuole biogenesis. J. Biol. Chem. 2000, 275, 24686–24692.
- Luo, H.R.; Huang, Y.E.; Chen, J.C.; Saiardi, A.; Iijima, M.; Ye, K.; Huang, Y.; Nagata, E.; Devreotes, P.; Snyder, S.H. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-ptdIns(3,4,5)P3 interactions. Cell 2003, 114, 559–572.
- Saiardi, A.; Nagata, E.; Luo, H.R.; Snowman, A.M.; Snyder, S.H. Identification and characterization of a novel inositol hexakisphosphate kinase. J. Biol. Chem. 2001, 276, 39179–39185.
- Chakraborty, A. The inositol pyrophosphate pathway in health and diseases. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1203–1227.
- Szijgyarto, Z.; Garedew, A.; Azevedo, C.; Saiardi, A. Influence of inositol pyrophosphates on cellular energy dynamics. Science 2011, 334, 802–805.
- Saiardi, A.; Bhandari, R.; Resnick, A.C.; Snowman, A.M.; Snyder, S.H. Phosphorylation of proteins by inositol pyrophosphates. Science 2004, 306, 2101–2105.
- Takazawa, K.; Perret, J.; Dumont, J.E.; Erneux, C. Molecular cloning and expression of a human brain inositol 1,4,5-trisphosphate 3-kinase. Biochem. Biophys. Res. Commun. 1991, 174, 529–535.
- Fridy, P.C.; Otto, J.C.; Dollins, D.E.; York, J.D. Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J. Biol. Chem. 2007, 282, 30754–30762.
- Azevedo, C.; Szijgyarto, Z.; Saiardi, A. The signaling role of inositol hexakisphosphate kinases (IP6Ks). Adv. Enzym. Regul. 2011, 51, 74–82.
- Safrany, S.T.; Caffrey, J.J.; Yang, X.; Bembenek, M.E.; Moyer, M.B.; Burkhart, W.A.; Shears, S.B. A novel context for the ‘MutT’ module, a guardian of cell integrity, in a diphosphoinositol polyphosphate phosphohydrolase. EMBO J. 1998, 17, 6599–6607.
- Onnebo, S.M.; Saiardi, A. Inositol pyrophosphates modulate hydrogen peroxide signalling. Biochem. J. 2009, 423, 109–118.
- Barker, C.J.; Wright, J.; Hughes, P.J.; Kirk, C.J.; Michell, R.H. Complex changes in cellular inositol phosphate complement accompany transit through the cell cycle. Biochem. J. 2004, 380, 465–473.
- Wundenberg, T.; Grabinski, N.; Lin, H.; Mayr, G.W. Discovery of InsP6-kinases as InsP6-dephosphorylating enzymes provides a new mechanism of cytosolic InsP6 degradation driven by the cellular ATP/ADP ratio. Biochem. J. 2014, 462, 173–184.
- Bennett, M.; Onnebo, S.M.N.; Azevedo, C.; Saiardi, A. Inositol pyrophosphates: Metabolism and signaling. Cell. Mol. Life. Sci. 2006, 63, 552–564.
- Desfougères, Y.; Wilson, M.S.C.; Laha, D.; Miller, G.J.; Saiardi, A. ITPK1 mediates the lipid-independent synthesis of inositol phosphates controlled by metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 24551–24561.
- Rao, F.; Cha, J.; Xu, J.; Xu, R.; Vandiver, M.S.; Tyagi, R.; Tokhunts, R.; Koldobskiy, M.A.; Fu, C.; Barrow, R.; et al. Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2. Mol. Cell 2014, 54, 119–132.
- Long, Y.C.; Cheng, Z.; Copps, K.D.; White, M.F. 2011. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol. Cell. Biol. 2011, 31, 430–441.
- Osaki, M.; Oshimura, M.; Ito, H. The PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676.
- Currie, R.A.; Walker, K.S.; Gray, A.; Deak, M.; Casamayor, A.; Downes, C.P.; Cohen, P.; Alessi, D.R.; Lucocq, J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide- dependent protein kinase-1. Biochem. J. 1999, 337, 575–583.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6.
- MacKenzie, R.W.; Elliott, B.T. Akt/PKB activation and insulin signaling: A novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab. Syndr. Obes. 2014, 7, 55–64.
- Zhang, Z.; Liu, H.; Liu, J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract. 2019, 156, 107092.
- Xie, Z.; Dong, Y.; Zhang, J.; Scholz, R.; Neumann, D.; Zou, M.H. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol. Cell. Biol. 2009, 29, 3582–3596.
- Zhu, Q.; Ghoshal, S.; Rodrigues, A.; Gao, S.; Asterian, A.; Kamenecka, T.M.; Barrow, J.C.; Chakraborty, A. Adipocyte-specific deletion of Ip6k1 reduces diet-induced obesity by enhancing AMPK-mediated thermogenesis. J. Clin. Investig. 2016, 126, 4273–4288.
- Kaidanovich, O.; Eldar-Finkelman, H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert Opin. Ther. Targets 2002, 6, 555–561.
- Izumiya, Y.; Hopkins, T.; Morris, C.; Sato, K.; Zeng, L.; Viereck, J.; Hamilton, J.A.; Ouchi, N.; LeBrasseur, N.K.; Walsh, K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008, 7, 159–172.
- Chakraborty, A.; Koldobskiy, M.A.; Bello, N.T.; Maxwell, M.; Potter, J.J.; Juluri, K.R.; Maag, D.; Kim, S.; Huang, A.S.; Dailey, M.J.; et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 2010, 143, 897–910.
- Barker, C.J.; Berggren, P.O. New horizons in cellular regulation by inositol polyphosphates: Insights from the pancreatic beta-cell. Pharmacol. Rev. 2013, 65, 641–669.
- Saiardi, A. How inositol pyrophosphates control cellular phosphate homeostasis? Adv. Biol. Regul. 2012, 52, 351–359.
- Illies, C.; Gromada, J.; Fiume, R.; Leibiger, B.; Yu, J.; Juhl, K.; Yang, S.N.; Barma, D.K.; Falck, J.R.; Saiardi, A.; et al. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science 2007, 318, 1299–1302.
- Morrison, B.H.; Bauer, J.A.; Hu, J.; Grane, R.W.; Ozdemir, A.M.; Chawla-Sarkar, M.; Gong, B.; Almasan, A.; Kalvakolanu, D.V.; Lindner, D.J. Inositol hexakisphosphate kinase 2 sensitizes ovarian carcinoma cells to multiple cancer therapeutics. Oncogene 2002, 21, 1882–1889.
- Nagata, E.; Luo, H.R.; Saiardi, A.; Bae, B.I.; Suzuki, N.; Snyder, S.H. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J. Biol. Chem. 2005, 280, 1634–1640.
- Nagata, E.; Saiardi, A.; Tsukamoto, H.; Okada, Y.; Itoh, Y.; Satoh, T.; Itoh, J.; Margolis, R.L.; Takizawa, S.; Sawa, A.; et al. Inositol hexakisphosphate kinases induce cell death in Huntington disease. J. Biol. Chem. 2011, 286, 26680–26686.
- Nagata, E.; Nonaka, T.; Moriya, Y.; Fujii, N.; Okada, Y.; Tsukamoto, H.; Itoh, J.; Okada, C.; Satoh, T.; Arai, T.; et al. Inositol hexakisphosphate kinase 2 promotes cell death in cells with cytoplasmic TDP-43 aggregation. Mol. Neurobiol. 2016, 53, 5377–5383.
- Chakraborty, A.; Koldobskiy, M.A.; Sixt, K.M.; Juluri, K.R.; Mustafa, A.K.; Snowman, A.M.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc. Natl. Acad. Sci. USA 2008, 105, 1134–1139.
- Koldobskiy, M.A.; Chakraborty, A.; Werner, J.K., Jr.; Snowman, A.M.; Juluri, K.R.; Vandiver, M.S.; Kim, S.; Heletz, S.; Snyder, S.H. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc. Natl. Acad. Sci. USA 2010, 107, 20947–20951.
- Chakraborty, A.; Kim, S.; Snyder, S. Inositol pyrophosphates as mammalian cell signals. Sci. Signal. 2011, 4, re1.
- Saiardi, A.; Resnick, A.C.; Snowman, A.M.; Wendland, B.; Snyder, S.H. Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinaserelated protein kinases. Proc. Natl. Acad. Sci. USA 2005, 102, 1911–1914.
- Morrison, B.H.; Tang, Z.; Jacobs, B.S.; Bauer, J.A.; Lindner, D.J. Apo2L/TRAIL induction and nuclear translocation of inositol hexakisphosphate kinase 2 during IFN-beta-induced apoptosis in ovarian carcinoma. Biochem. J. 2005, 385, 595–603.
- Rao, F.; Xu, J.; Fu, C.; Cha, J.Y.; Gadalla, M.M.; Xu, R.; Barrow, J.C.; Snyder, S.H. Inositol pyrophosphates promote tumor growth and metastasis by antagonizing liver kinase B1. Proc. Natl. Acad. Sci. USA 2015, 112, 1773–1778.
- Carretero, J.; Shimamura, T.; Rikova, K.; Jackson, A.L.; Wilkerson, M.D.; Borgman, C.L.; Buttarazzi, M.S.; Sanofsky, B.A.; McNamara, K.L.; Brandstetter, K.A.; et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 2010, 17, 547–559.
- Roy, B.C.; Kohno, T.; Iwakawa, R.; Moriguchi, T.; Kiyono, T.; Morishita, K.; Sanchez-Cespedes, M.; Akiyama, T.; Yokota, J. Involvement of LKB1 in epithelial-mesenchymal transition (EMT) of human lung cancer cells. Lung Cancer 2010, 70, 136–145.
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843.
- Song, P.; Xie, Z.; Wu, Y. Protein kinase Czeta-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J. Biol. Chem. 2008, 283, 12446–12455.
- Nilsson, J.; Helou, K.; Kovács, A.; Bendahl, P.O.; Bjursell, G.; Fernö, M.; Carlsson, P.; Kannius-Janson, M. Nuclear janus-activated kinase 2/nuclear factor 1-C2 suppresses tumorigenesis and epithelial-to-mesenchymal transition by repressing Forkhead box F1. Cancer Res. 2010, 70, 2020–2029.
- Jadav, R.S.; Kumar, D.; Buwa, N.; Ganguli, S.; Thampatty, S.R.; Balasubramanian, N.; Bhandari, R. Deletion of inositol hexakisphosphate kinase 1 (IP6K1) reduces cell migration and invasion, conferring protection from aerodigestive tract carcinoma in mice. Cell Signal. 2016, 28, 1124–1136.
- Grudzien-Nogalska, E.; Jiao, X.; Song, M.G.; Hart, R.P.; Kiledjian, M. Nudt3 is an mRNA decapping enzyme that modulates cell migration. RNA 2016, 22, 773–781.
- Morrison, B.H.; Haney, R.; Lamarre, E.; Drazba, J.; Prestwich, G.D.; Lindner, D.J. Gene deletion of inositol hexakisphosphate kinase 2 predisposes to aerodigestive tract carcinoma. Oncogene 2009, 28, 2383–2392.
- Morrison, B.H.; Bauer, J.A.; Kalvakolanu, D.V.; Lindner, D.J. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J. Biol. Chem. 2001, 276, 24965–24970.
- Aoki, M.; Sobek, V.; Maslyar, D.J.; Hecht, A.; Vogt, P.K. Oncogenic transformation by beta-catenin: Deletion analysis and characterization of selected target genes. Oncogene 2002, 21, 6983–6991.
- Zhang, Z.; Wang, Y.; Yao, R.; Li, J.; Lubet, R.A.; You, M. p53 Transgenic mice are highly susceptible to 4-nitroquinoline-1-oxide-induced oral cancer. Mol. Cancer Res. 2006, 4, 401–410.
- Barcellos-Hoff, M.H.; Cucinotta, F.A. New tricks for an old fox: Impact of TGFβ on the DNA damage response and genomic stability. Sci. Signal. 2014, 2014, 7.
- Zhu, Q.; Ghoshal, S.; Tyagi, R.; Chakraborty, A. Global IP6K1 deletion enhances temperature modulated energy expenditure which reduces carbohydrate and fat induced weight gain. Mol. Metab. 2017, 6, 73–85.
- Moritoh, Y.; Oka, M.; Yasuhara, Y.; Hozumi, H.; Iwachidow, K.; Fuse, H.; Tozawa, R. Inositol hexakisphosphate kinase 3 regulates metabolism and lifespan in mice. Sci. Rep. 2016, 6, 32072.
- Xu, Y.; Li, H.; Bajrami, B.; Kwak, H.; Cao, S.; Liu, P.; Zhou, J.; Zhou, Y.; Zhu, H.; Ye, K.; et al. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc. Natl. Acad. Sci. USA 2013, 110, 7726–7731.
- Malla, A.B.; Bhandari, R. IP6K1 is essential for chromatoid body formation and temporal regulation of TNP2 and PRM2 expression in mouse spermatids. J. Cell Sci. 2017, 130, 2854–2866.
- Worley, J.; Luo, X.; Capaldi, A.P. Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L. Cell. Rep. 2013, 3, 1476–1482.
- Brehm, M.A.; Wundenberg, T.; Williams, J.; Mayr, G.W.; Shears, S.B. A non-catalytic role for inositol 1,3,4,5,6-pentakisphosphate 2-kinase in the synthesis of ribosomal RNA. J. Cell Sci. 2013, 126, 437–444.
- Kim, S.; Kim, S.F.; Maag, D.; Maxwell, M.J.; Resnick, A.C.; Juluri, K.R.; Chakraborty, A.; Koldobskiy, M.A.; Cha, S.H.; Barrow, R.; et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011, 13, 215–221.
- Nair, V.S.; Gu, C.; Janoshazi, A.K.; Jessen, H.J.; Wang, H.; Shears, S.B. Inositol pyrophosphate synthesis by diphosphoinositol pentakisphosphate kinase-1 is regulated by phosphatidylinositol(4,5)bisphosphate. Biosci. Rep. 2018, 38.
- Padmanabhan, U.; Dollins, D.E.; Fridy, P.C.; York, J.D.; Downes, C.P. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: Use in defining biological roles and metabolic relationships of inositol pyrophosphates. J. Biol. Chem. 2009, 284, 10571–10582.
- Sarmah, B.; Wente, S.R. Inositol hexakisphosphate kinase-2 acts as an effector of the vertebrate Hedgehog pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 19921–19926.
- Ghoshal, S.; Zhu, Q.; Asteian, A.; Lin, H.; Xu, H.; Ernst, G.; Barrow, J.C.; Xu, B.; Cameron, M.D.; Kamenecka, T.M.; et al. TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol. Metab. 2016, 5, 903–917.
- Chang, Y.T.; Choi, G.; Bae, Y.S.; Burdett, M.; Moon, H.S.; Lee, J.W.; Gray, N.S.; Schultz, P.G.; Meijer, L.; Chung, S.K.; et al. Purine-based inhibitors of inositol-1,4,5-trisphosphate-3-kinase. Chembiochem 2002, 3, 897–901.
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641.
- Vincent, E.E.; Elder, D.J.; Thomas, E.C.; Phillips, L.; Morgan, C.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavare, J.M. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761.
- Mundi, P.S.; Sachdev, J.; Mccourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956.


