 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joanna Zawacka | + 4666 word(s) | 4666 | 2020-09-29 06:07:10 | | | |
| 2 | Joanna Zawacka | Meta information modification | 4666 | 2020-10-06 22:37:59 | | | | |
| 3 | Bruce Ren | Meta information modification | 4666 | 2020-10-09 07:45:27 | | | | |
| 4 | Joanna Zawacka | Meta information modification | 4666 | 2020-10-13 12:38:03 | | | | |
| 5 | Bruce Ren | Meta information modification | 4666 | 2020-10-21 09:53:53 | | | | |
| 6 | Bruce Ren | Meta information modification | 4666 | 2020-10-21 09:55:52 | | |
Video Upload Options
p53 and p73 are critical tumor suppressors that are often inactivated in human cancers through various mechanisms. Owing to their high structural homology, the proteins have many joined functions and recognize the same set of genes involved in apoptosis and cell cycle regulation. p53 is known as the ‘guardian of the genome’ and together with p73 forms a barrier against cancer development and progression. The TP53 is mutated in more than 50% of all human cancers and the germline mutations in TP53 predispose to the early onset of multiple tumors in Li–Fraumeni syndrome (LFS), the inherited cancer predisposition. In cancers where TP53 gene is intact, p53 is degraded. Despite the ongoing efforts, the treatment of cancers remains challenging. Presently, the endeavors focus on reactivating p53 exclusively, neglecting the potential of the restoration of p73 protein for cancer eradication. Taken that several small molecules reactivating p53 failed in clinical trials, there is a need to develop new treatments targeting p53 proteins in cancer. This review outlines the most advanced strategies to reactivate p53 and p73 and describes drug repurposing approaches for the efficient reinstatement of the p53 proteins for cancer therapy.
1. Introduction
It is the media hype and the unreasonable costs of the majority of new cancer treatments, often delivering only a marginal benefit, which harm cancer patients. More often than not, new treatments fail to deliver advancement in the outcomes, including overall survival. Surrogate endpoints applied in clinical trials usually include disease-free survival (DFS) (or progression-free survival), or overall response rates (ORR) as the primary outcome instead of a patient-centered, overall survival (OS). This, together with the underreported financial conflicts of interest among the decisive bodies, and the biased selection criteria for clinical trial randomization, all lead to the accelerated approval of expensive treatments which only marginally improve the patients’ outcome. As the situation looks now, it leaves little or no room for the introduction of the unbiased approach in cancer care, as illustrated by Vinayak Prasad in the book Malignant [1].
One way to overcome the burden of the skyrocketing costs of treatments of questionable benefit to patients is to apply a drug repurposing approach. Drug repurposing uses an existing drug for a different medical indication. In oncology, hard drug repurposing conveys the application of the drug from the non-oncology application, to improve the outcome of cancer therapy, often at a much lower cost than that of bringing a new treatment to the market [2]. This approach is economical as it takes advantage of the clinical information that is already available for the given drug, such as pharmacokinetic and pharmacodynamic profiles, maximum tolerated dose or clinical safety profile, which allows for shorter times for the treatment’s implementation into practice [3].
p53 and its ancestor family members, p73 and p63, evolved in the multicellular organisms as the sensors of the DNA damage. All proteins constitute a critical barrier against cancer development in humans and regulate the expression of genes involved in apoptosis, cell cycle regulation or DNA repair. p53 is the most commonly inactivated protein in human cancers, either due to the mutations in its gene promoting the loss of wild-type (wt) p53 function, or due to the overactivated oncogenic inhibitors, like MDM2 and/or MDMX [4]. p53 works together with p73, a p53 protein family member, which also includes p63. p73 evolved earlier than p53 in vertebrates and all three proteins share a similar sequence, architecture, and function. The structure–function similarity among the p53 protein family allows us to assume that small molecules activating p53 will also work on p73, which is discussed in Sections 2 and 3.
In Li–Fraumeni syndrome, the inherited cancer predisposition, TP53 mutations have high penetrance, and the loss of p53 function drives the early onset of multiple tumors. The germline TP53 mutations make the treatment of LFS patients challenging due to the genotoxicity of currently available therapies, enhancing the probability of the development of secondary malignancies. Thus, the LFS patients are in majority treated with surgery before implementing chemotherapy or radiotherapy. Some hopes for improved therapy of tumors in LFS patients are seen with immunotherapies [5]. However, the cost of immunotherapies and other non-genotoxic modalities e.g., the CAR-T therapy (app. USD 2 million with accompanying costs), calls for urgent development of new, more affordable treatments for cancer patients with the mutated p53 [6].
Taken the abovementioned issues, the critical role of p53 in cancer initiation and progression, and the recently reported failure of the promising MDM2 inhibitors, RG7112, and idasanutlin in clinical trials, there is a need for enhanced efforts into development of therapies reactivating the p53 protein family [7]. This review describes structures and tumor suppressor functions of p53 and p73, selected approaches to reactivate p53 proteins’ function in tumors and highlights the potential of drug repurposing approach for restoration of p53 and p73 for cancer therapy.
2. Structure and Tumor Suppressor Function of p53
2.1. p53
p53 is a protein of the domain structure and a transcription factor binding specifically to DNA consensus sequence consisting of two consecutive half-sites as a tetramer [8]. p53 is known to undergo multiple post-translational modifications including phosphorylation, ubiquitination, sumoylation, neddylation, acetylation, methylation, or recently described UFMylation [9], which are necessary for p53 cellular turnover. In non-stressed cells, the half-life of p53 is around 20 min and the protein becomes stabilized and activated by the cascade of events provoked by cellular stress signals (reviewed in [10]). Stabilization of p53 is achieved by the decrease in the affinity of MDM2 to p53 (or HDM2 in humans), a major p53 E3 ubiquitin ligase which drives p53 for proteasomal degradation, in the cytosol and in the nucleus [11][12]. Activation of p53 transcription function occurs upon the inhibition of the binding of MDM2 to the N-terminal domain of p53 at the target DNA sequence. Since MDM2 is also a p53 target gene, a negative feedback loop exists that regulates p53 activity (Figure 1) [11].
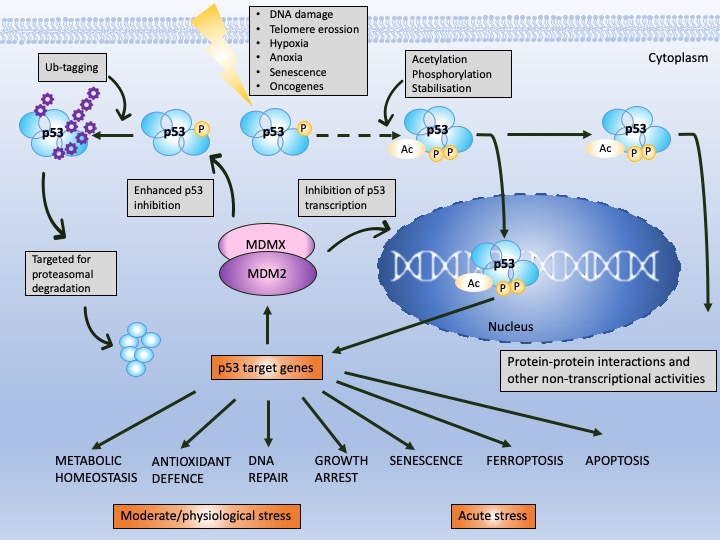
Figure 1. p53 and MDM2 as a hub of p53-dependent cellular responses—a simplified model. Under physiological conditions, p53 is degraded by MDM2, E3 ubiquitin ligase, which, depending on the level of cellular stress, can have either high or low affinity to p53. MDM2 is responsible for p53 monoubiquitination (driving p53 nuclear export) and polyubiquitination of p53 (driving p53 ubiquitin-dependent proteasomal degradation) and prevents p53 acetylation and transcriptional activation by p300 acetyltransferase. The affinity of MDM2 to p53 is enhanced upon hetero-dimerization with its homolog, MDMX protein. Upon stress, p53 undergoes phosphorylation and acetylation (the sites depend on the type and severity of stress) and recognizes its target genes. MDM2 and MDMX may prevent p53 from initiating the transcription through direct binding, which hinders the binding of the transcriptional co-activators. The sets of the target genes that become activated/repressed by p53 are often interrelated. In addition to transcription-dependent activity, cytoplasmic p53 functions through protein–protein interactions to modulate apoptosis, miRNA maturation or the repair of double-strand breaks (DBS). The dotted line represents a multistep process. Adapted from Levine, 2020 [13], Levine and Oren, 2009 [14] and Bode and Dong, 2004 [15].
Next, MDM2 protein, at mild stress, monoubiqutinates p53, enforcing its nuclear export and enabling p53 non-transcriptional activity. MDM2 activity towards p53 is enhanced by its homolog, MDMX protein, which lacks the E3 ligase activity but binds to the N-terminus of p53 and MDM2 alike, inhibiting its transcription function [16]. Apart from MDM2, other ligases play a role in altering p53 stability like Trim family members or Pirh or bacterial or viral proteins, such as SV40 or E6 protein of the HPV virus [17]. It is, however, the MDM2–p53 hub that is responsible for regulating multiple cellular processes in human cells, such as apoptosis, cell cycle, DNA repair, antioxidant response or senescence as well as metabolism (Figure 1). Furthermore, p53 also regulates ferroptosis, iron-related cell death and has the transcription-independent function in apoptosis (binding to Bcl2-family of proteins), in miRNA maturation (binding to Drosha-complex proteins) and in DNA repair [18]. Its pivotal role is to orchestrate the response to genotoxic, oxidative, and oncogene-induced stress [19].
In response to mild DNA damage, activation of p53 transcription initiates cell cycle inhibition, necessary for the DNA repair to occur, and both processes converge on a cascade of protein–protein interactions (PPIs) [20][21]. If the DNA damage cannot be repaired, the cell is directed to apoptosis, a programmed cell death. In that case, p53 transactivates BCL2-associated X, apoptosis regulator (BAX), p53 upregulated modulator of apoptosis (PUMA; also known as BBC3) and NOXA (also known as PMAIP1) [22] or interacts directly with the multidomain anti-apoptotic (Bcl-xL and Bcl-2) and proapoptotic (Bak) Bcl-2 members at mitochondria and induces mitochondrial outer membrane permeabilization and consequent cytochrome c release and apoptosis (reviewed in [23]).
2.1.1. p53 Structure
The N-terminus domain of p53 includes transactivation domain 1 (TAD1, depicted as T1) and TAD2 (T2) (Figure 2A, upper panel). TAD1 and 2 work synergistically to induce transcription and are sites of phosphorylation events leading to inhibition of MDM2-p53 complex and to activation of p53-dependent response (reviewed in [24]). The X-ray structure of the MDM2 N-terminus and p53 N-terminal peptide complex shows that the minimal requirements for p53 to bind MDM2 are residues F19S20D21L22W23K24L25L26 [25][26]. p53 residues F19, W23 and L26 are responsible for binding with MDM2 and MDMX [27]. Taking into account the well-known structure of the MDM2-p53 complex, and the fact that the inhibition of the wild-type (wt) p53 via p53/MDM2/MDMX axis is essential for cancer to develop (reviewed in [4]), inhibition of the MDM2-p53 and MDMX-p53 interactions has become a very promising strategy for cancer therapy and is described in more detail below. The TAD domain of p53 is rendered unfolded and adapts a transiently stable secondary structure. In particular, the region from Phe19 to Leu22, responsible for binding to MDM2 protein, exhibits local helix propensity [28] and is sensitive to the charge-induced shifts. Interestingly, the liable p53 N-terminus can be targeted with small molecules that move the local charge and disrupt the helix. This prevents MDM2-p53 interactions as demonstrated for a small molecule RITA, a compound that affects the interaction between p53 and MDM2 through the change in conformation of p53 N-terminus [29][30]. Since this phenomenon is not fully understood yet, it will not be discussed in this review.
p53 binds specifically to its consensus DNA sequence through the DNA binding domain. The DNA binding domain (DBD) located centrally, spans the amino acids from 98 to 292, is preceded with the proline-rich region and two transactivation domains, TAD1 and TAD2 (Figure 2A, upper panel). The DBD domain is enriched in cysteine residues and contains an antiparallel β-sheet sandwich supported by loops L1, L2, and L3. Loops L2 and L3 contain amino acids for a tetrahedrally coordinated Zn2+ ion. The wild-type p53 protein recognizes the canonical DNA sequence motif by binding to DNA through residues K120, R273, A276, C277, R280, R283 and residues S241 and R248, which are located at the ends of two β-sheets [31]. Since the DBD is important for p53-facilitated transcription, it is a site of multiple inactivating mutations which are found in cancer.
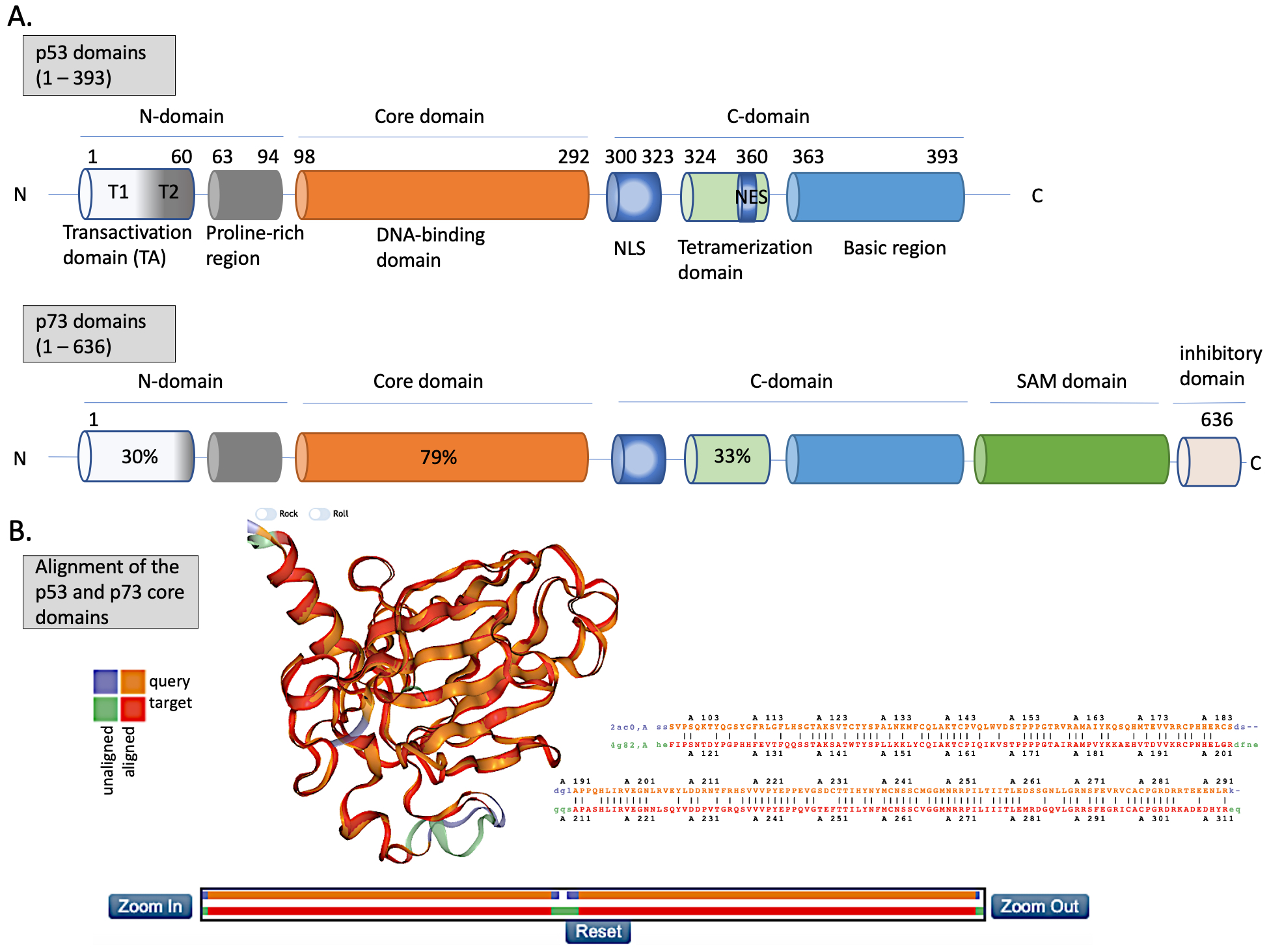
Figure 2. The structures of p53 and p73. (A) Upper panel—domains in p53 protein. Lower panel—percentage homology of residues between p53 and p73 are presented and the values are indicated for each individual structural domain. T1, T2—transactivation domain (TAD) 1 and 2; NLS—nuclear localization signal; NES—nuclear export signal; SAM—sterile alpha-motif. Adapted from Tanaka et al. [32], Joanna Zawacka-Pankau et al. [33] and Melino et al., [34]. (B) Structure alignment of p53 core domain (PDB ID 2AC0 [8]) and p73 core domain (PDB ID 4G82 [35]) generated using Top Match Services with opacity of unmatched pairs of 0.7. https://topmatch.services.came.sbg.ac.at/index_ngl.html [36]. At the C-terminus, the regulatory basic domain is located [37] which is involved in the interactions with DNA through non-specific DNA binding allowing for distinctive target gene recognition by p53 and p73 [38].
2.1.2. p53 Inactivation in Cancer
p53 is activated in response to oncogene-induced stress (Figure 1) and is therefore the most commonly mutated gene in cancer. More than 50% all of human cancers harbor the inactivating mutations and the six most common are the missense mutations hindering the activity of DBD domain: R175, G245, R248, R249, R273, R282 ([39][40] https://p53.iarc.fr/). The mutations render p53 inactive and/or promote the gain of new functions [41]. Studies demonstrated the feasibility of reactivating mutant p53 with small molecules (reviewed in [42]) and an advanced clinical example is described below. In cases in which TP53 gene remains intact, p53 protein is degraded by the upregulated or hyperactive MDM2 protein, which acts in concert with MDMX (reviewed in [4][13]) (Figure 1). MDM2 was found to be overexpressed in many tumor types via several mechanisms including gene amplification or enhanced transcription [43]. MDM2 is amplified in sarcomas, bladder cancer or glioblastoma (https://www.cbioportal.org/). The protein is expressed from two promoters [44] and the single nucleotide polymorphisms (SNP), SNP309G-allele and SNP55T-allele in promoter 2 of MDM2 were described to enhance the binding of Sp1 transcription factor and to increase MDM2 expression. Accumulated MDM2 promotes p53 downregulation in several human cancers [45][46][47]. Next, MDM2 is overactivated in cancers because of the inhibition of p14ARF tumor suppressor. In normal cells, oncogene activation stimulates p53 stabilization due to activation of p14ARF. p14ARF binds to MDM2 and induces the nucleolar import of MDM2 protein. The binding of p14ARF prevents MDM2-mediated transactivational silencing of p53 and p53 degradation [48][49]. p14ARF was reported to be inhibited in cancer cell lines and tumor tissues through the INK4a/ARF locus deletion or promoter hypermethylation, and the homozygous deletions of p14ARF have prognostic significance in cancer [50][51][52]. Similarly to ARF, MDM2 is also negatively regulated by the ribosomal proteins (RP) L11 and RPL5 [53] that are activated by ribosomal or nucleor stress.
The growing evidence implies that the mechanisms leading to p53 inactivation in cancer, to some extent, also apply to p73, a p53 protein family member. For example, p73 is activated by RBL11 and RBL5 in cancers [54]. This will be discussed in more detail in Section 2.3.
2.1.3. Pharmacological Reactivation of p53
The most advanced mutant p53 reactivating compound is APR-246 (known as eprenetapopt) discovered by Klas Wiman and colleagues [55]. APR-246 is converted to methyl quinuclidinone (MQ) and acts as Michael acceptor which targets specific cysteine residues in p53 core domain [56][57]. The binding of MQ to cysteine 277 increases the thermostability of the core domain in vitro and cysteine 124 and 277 are crucial for reactivation of mutp53-R175H in cancer cells. APR-246 also inhibits thioredoxin reductase (TrxR) and binds to glutathione which boosts accumulation of reactive oxygen species and contributes to cancer cells’ death (reviewed in [58]). The compound is studied in Phase III clinical trial in combination with azacytidine in TP53 mutated myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) (reviewed in [59]).
Extensive studies led to the development of rationally designed small-molecule inhibitors, nutlins, that bind the MDM2 hydrophobic pocket with high affinity, and efficiently outcompete p53 from the binding site [60]. The pivotal study with nutlin-3 (IUPAC: 4-[(4S,5R)-4,5-bis(4-chlorophenyl)-2-(4-methoxy-2-propan-2-yloxyphenyl)-4,5-dihydroimidazole-1-carbonyl]piperazin-2-one), showed that it mimics the three key interactions of p53. Specifically, the imidazoline fits into the MDM2 binding site protruding three hydrophobic groups into subpockets that are normally occupied by the p53 Phe19, Trp23, and Leu26 residues and the piperazine ring attached to the N1 of the imidazoline is outside the binding site and does not contact MDM2. Nutlin has a much lower affinity to MDMX [61][62] and thus, is ineffective in tumors that overexpress both MDM2 and MDMX [63]. Similarly to MDM2, p53 regulates MDMX as it binds to mRNA of MDMX and regulates its translation. More specifically, the p53 DBD domain binds the 5′ untranslated region (UTR) of the MDMX mRNA in a zinc-dependent manner and through the partaking of the p53 N-terminus controls MDMX synthesis generating a negative feedback loop between p53 and MDMX as is the case for MDM2 [64]. The initial success of nutlin-3 commenced the development of a series of potent MDM2-p53 inhibitors and their extensive testing in the clinical setting. However, recently the failure of highly specific MDM2 inhibitors, RG7112 and idasanutlin in clinical trials was reported. Yet, a compound called APG-115, an oral MDM2 inhibitor of high affinity, was tested in combination with KEYTRUDA® in a Phase Ib/II trial [65]. Further studies will show the clinical efficacy of the drug.
One of the promising strategies to treat cancers with wtp53 is to apply dual inhibitors of MDM2-p53 and MDMX-p53 interactions [63]. The most advanced examples of such approach are stapled peptides, α-helical p53 stapled peptidomimetics among which the ALRN-6924 peptide is the only one in early phase clinical development [66]. Small-molecule, dual antagonists have not yet been tested in the clinical setting and thus, new approaches allowing for rapid translation into clinical practice are needed.
An emerging strategy to target tumors with inactive p53 is to reactivate other p53 protein family members. p73 is an important tumor suppressor, rarely mutated in cancer. The accumulated published data imply that p73, when reactivated, compensates for p53 loss and induces apoptosis and tumor regression in vivo, as discussed in detail below.
2.2. p73
Since its discovery in 1997, p73 has been intensively studied because of its high structural similarity to p53 and owning to the possibility to compensate for p53 loss in tumors [67]. p73 has higher than p63 percentage of the homology in the DNA binding domain to p53 (Figure 2A,B (lower panel)) and forms open tetramers in a manner similar to p53, while p63 forms two closed dimers [68][69]. Such similarity to p53 allows making an assumption that p73 might recognize and activate many of p53 target genes and that similar pharmacological approaches can be employed to activate p73 protein for cancer therapy. Taking into consideration the difference in structure and the limited data regarding p63 reactivation for cancer therapy, this review will focus on p53 and p73 solely.
2.2.1. p73 Structure
p73 is expressed in several isoforms that have distinct functions. The two major p73 isoforms dictating the cell fate upon cellular stress and chemotherapy treatment are TA isoforms and DN isoforms. p73 has two promoters—P1 in the 5′ untranslated region upstream of the noncoding exon 1, and P2 within the 23 kb spanning intron 3, triggering the synthesis of two distinct isoforms (reviewed in [70]). TA isoforms are transcriptionally active and act as tumor suppressors and DN isoforms, which lack the N-terminus, arise in cells through the alternative promoter usage of P2 and through the alternative splicing. Importantly, when the ratio between the isoforms is altered due to, e.g., the methylation of CpG islands in promoter 1, DN isoforms accumulate and can interact with and inhibit TA isoforms and p53 [71]. In addition to inhibiting p53 and p73, ΔNp73 has other oncogenic functions such as binding to HIF1a and promoting its stability and tumor metastasis [72], driving chemoresistance by regulating the expression of the multi-drug resistance genes ABCB1 and 5 [73], interacting with TGFβ signaling by inducing its target genes PAI-1 and Col1a1 [74], or inhibiting PTEN tumor suppressor [75][76]. Next, the alternative splicing at the C-terminus generates the C-terminal isomeric forms of p73, which are expressed both in healthy and in cancer cells. The longest isoform, TAp73α, contains a highly conserved sterile motif (SAM) (Figure 2A (lower panel)), which is a protein–protein interaction module (reviewed in [34]). In total, there are 35 isoforms of p73, which adds complexity while studying p73 biology [77].
Structural homology between the DBD domains (Figure 2B) explains why p53 and p73 transactivate many of the same target genes, such as PUMA, CDKN1A, or BAX. Similarly to p53, p73 maintains the tumor suppressor function by guarding the genomic stability and driving cell cycle arrest, replicative senescence or apoptosis [78][79]. Reports also point to the involvement of p73 in regulating metabolism [80]. p73 activity is coordinated by a plethora of post-translational modifications, such as ubiquitination, phosphorylation, acetylation, or sumoylation driven by oncogenic insult or IR-mediated DNA damage [81]. Like p53, p73 transcription is inhibited by binding to MDM2 [82] and MDMX [83] through its TAD domain (Figure 3). The affinities of MDM2 and MDMX to p73 are of the same order as to p53, Kd (μM) = 1.4 and Kd (μM) = 0.22, respectively [84]. Thus, one can conclude that there is a high structural and functional similarity between the domains of p53 and p73. The similarity between the proteins was shown by molecular dynamics simulations which described similar, transient structural fluctuations of the p53 and the p73 α-helixes when in proximity to the MDM2 binding pocket [85].
p73, like p53, has both transcription-dependent and independent functions. Transcription activity of the longest form of p73, TAp73α, similarly to p53, is induced by acetylation by p300 and CREB-binding protein (CBP) acetyltransferases [86]. Next, p73 transcriptional activity and p73-driven cell death are significantly enhanced by YAP (YES-associated protein) through p300/CBP. On the other hand, YAP stability is increased by DNA damage via c-Abl kinase-mediated phosphorylation promoting the reinforced p73-mediated apoptosis. C-Abl is activated by DNA damage and is known to activate p53 [87][88]. In addition, p73 is directly phosphorylated by c-Abl at Tyr99 which further increases its transcriptional activity and enhances DNA repair driven by TAp73 [89]. In addition to promoting p73 transcription activity, YAP also outcompetes MDM2 and ITCH E3-ligase from the complex with p73, promoting TAp73 protein stability [90].
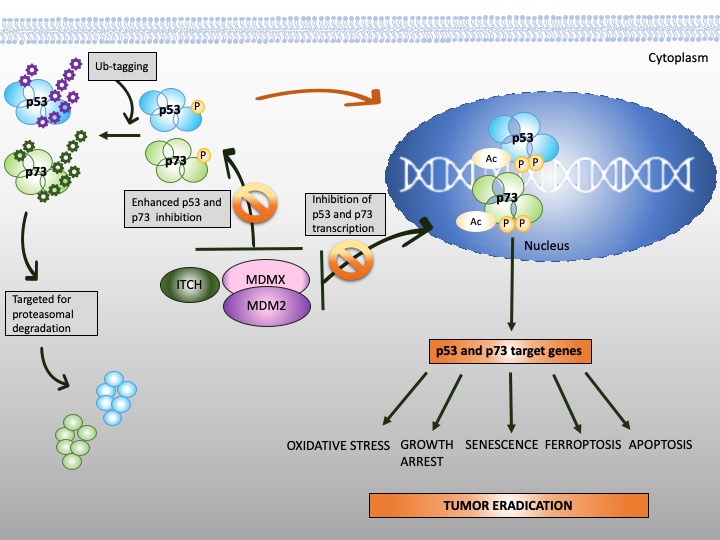
Figure 3. Reinstatement of p53 and p73 to treat cancer. Both p53 and p73 are rendered inactive in tumor cells through enhanced ubiquitination by MDM2/MDMX and MDM2/MDMX/ITCH axis, respectively. In addition to enhanced protein degradation, the transcriptional activity of p53 and p73 is inhibited through binding to MDM2 and MDMX. Targeting protein-protein interactions with small molecules or peptidomimetics (orange, crossed circles) stabilizes p53 and p73 and restores their transcription function (orange arrow). This, in turn, promotes tumor eradication through multiple mechanisms, as depicted in the scheme.
The stability of p73 is mediated by E3 ubiquitin ligase. The major E3 ubiquitin ligase of p73 is HECT ligase ITCH [91]. MDM2 and MDMX both bind to N-terminus of p73 and inhibit its transcriptional activity [92][93]. Recent studies indicated that MDM2 promotes p73 proteolytic disassembly through interacting with ITCH [94][95] and that at high levels, MDM2 polyubiquitinates p73 and regulates p73-facilitated apoptosis [95]. p73 has also cytoplasmic, transcription-independent functions and after DNA damage induces apoptosis through noncanonical binding to anti-apoptotic Bcl-XL [96].
2.2.2. p73 Tumor Suppressor Function
After its discovery, the function of p73 in cancer was largely unexplored. Early studies demonstrated that the knockout of Tp53 leads to tumor development in mice [97]. The mice heterozygous for Tp73 (p73+/−) are tumor-prone[98], and the studies from the Tak Mak’s Lab demonstrated unequivocally that the knockout of TAp73 (TAp73−/−) leads to tumor development and infertility in vivo [99]. Around 70% of the mice cohort developed lung cancer, and the rest showed premature aging, which was attributed to the de-regulated metabolism. In these mice, infertility was a result of genomic instability. Aberrancy in the DNA repair system in TAp73−/− mice might affect the quality of oocytes in a manner similar to the one occurring during healthy aging and thus, may explain the observed phenotype. This study demonstrated for the first time that TAp73 is a powerful tumor suppressor involved in DNA repair. Next, Elsa Flore’s Lab showed that acute genetic depletion of ΔN isoforms of p73 induced regression of tumors developed in the Tp53-null background in vivo [100]. The mechanism of tumor regression was via the induction of apoptosis. Altogether, deletion of ∆Np73 compensates for p53 loss and this occurs through the upregulation of TAp73 and induction of apoptosis. Another study showed that depletion of MDM2-/- in Tp53-/- null tumors leads to the upregulation of p73, apoptosis and tumor regression via activated p73 [101]. Thus, the above-mentioned studies and others [102] comprise a large body of evidence that demonstrates that the deregulated p73 contributes to cancer development and progression and that accumulated TAp73 compensates for p53 loss and induces tumor suppression.
2.2.3. Pharmacological Reactivation of p73
Unlike TP53, TP73 gene is infrequently mutated in cancers [103]. Due to promoter hypomethylation, the oncogenic ΔNp73 isoform is upregulated in several cancers, including gastric, esophageal, thyroid and head and neck cancer and the cancers of the lung, breast or ovary. High ΔNp73 is linked to poor prognosis and treatment resistance [104][105]. One way to overcome oncogenic ΔNp73 is to alter the ratio between the isoforms by elevating the levels of TAp73. Whether pharmacological activation of TAp73 isoform can compensate for p53 loss has been controversial for a long time. Apart from IR-induced DNA damage, only a few molecules were described to directly or indirectly activate TAp73 in cancers [106]. Several pathways lead to inactivation of TAp73 in cancer. Firstly, it is the epigenetic modification at P1 and P2 which alters the ratio between TA/ΔN isoforms and next the binding to oncogenic protein inhibitors like ΔN isoforms, mutant p53 or MDM2 and MDMX. Thus, current efforts aim at direct or indirect targeting of protein–protein interactions to reactivate TAp73 in tumors.
A study using siRNA-mediated inhibition of ITCH demonstrated that cancer cells lacking p53 are more sensitive to ITCH silencing after treatment with chemotherapeutics and undergo rapid apoptosis due to p73 activation [107]. Hence, targeting ITCH-p73 interactions emerges as a promising approach for cancer therapy, which is discussed in Section 3. In addition to ITCH, p73, like p53, is subject to similar regulation by MDM2 protein. Cumulated evidence showed that at higher dose, Nutlin-3, the MDM2-p53 antagonist, induces TAp73 and apoptosis in cancer cells [108]. Furthermore, small molecule RETRA was described to target mutp53-p73 complex and to specifically suppress the growth of mutant p53-bearing tumor cells in vitro and in mouse xenografts [109]. Yet, the p73 and c-Abl kinase axis was described to significantly contribute to cisplatin-induced cytotoxicity in cancer cells with wtp53 [110]. Interestingly, another study showed that ΔNp63 mediates p73-dependent sensitivity to chemotherapy in triple-negative breast cancer [111]. Briefly, ΔNp63 promoted the survival of breast cancer cells by binding to TAp73 and inhibiting its proapoptotic activity, whereas breast cancer cells expressing ΔNp63a and high TAp73 exhibited cisplatin sensitivity that was dependent on TAp73. In response to treatment with cisplatin, TAp73 underwent c-Abl-dependent phosphorylation, which promoted dissociation of TAp73 from the complex with ΔNp63 and this triggered TAp73-dependent transcription of proapoptotic Bcl-2 family members and apoptosis. Next, a recent study showed that hypermethylation of P1 of TP73 gene correlates with the decrease in TAp73 and shorter overall survival of bladder cancer patients. A DNA demethylating agent, decitabine, decreased the methylation of CpGs in P1 of TP73 and increased the sensitivity to cisplatin in cell culture conditions [112]. The study from Christian Gaiddon’s Lab showed that HDAC significantly induces mRNA and protein levels of p73 and protein levels of p53 in gastric cancer cell lines after cisplatin treatment. This leads to the efficient induction of the proapoptotic genes PMAIP1 (NOXA) and BIK [113]. These findings support the key role of TAp73 in eliminating cancer cells in response to cisplatin and delineate p73 as a vital target of the drug.
In addition to the extended studies on cisplatin, p73 also sensitizes p53-null colon cancer cells (HCT 116 p53−/−) to withaferin A (WA), a plant-derived proteasomal inhibitor. WA stabilizes and activates TAp73 through the c-Jun N-terminal kinases - NAD(P)H dehydrogenase [quinone] 1 (JNK-NQO1) axis and reactive oxygen species-mediated response. In more detail, the study showed that WA induces p73 phosphorylation by JNK kinase, releases p73 from MDM2, stabilizes p73 on the protein level, and induces p73-dependent apoptosis in p53-null cells [114]. Next, a study with bortezomib (VelcadeÒ), a known proteasomal inhibitor approved by the FDA as a frontline treatment in Relapsed/Refractory multiple myeloma, further confirmed the ‘druggability’ of p73. Here, researchers used a pair of isogenic HCT 116 human colon cancer cell lines differing only in p53 status and showed that bortezomib induces TAp73 and apoptosis in cells lacking p53 [115]. These studies too, supported the notion that p73 can be targeted with small molecules and efficiently compensates for p53 loss in tumor suppression. Thus, based on the successful reports highlighted above, the strategy aiming at the targeted restoration of TAp73 for cancer therapy is feasible, and p73 is a promising therapeutic target in cancers.
References
- Prasad, V.K. Malignant: How Bad Policy and Bad Evidence Harm People with Cancer; Johns Hopkins University Press, Baltimore, 2020.
- Pantziarka, P.; André, N. Editorial: Drug Repurposing. Front. Med. 2019, 6, 154.
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58.
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies-a clinical synopsis. Cell Death Dis. 2020, 11, 237.
- Chen, L.; Xu, B.; Long, X.; Gu, J.; Lou, Y.; Wang, D.; Cao, Y.; Wang, N.; Li, C.; Wang, G.; et al. CAR T-cell therapy for a relapsed/refractory acute B-cell lymphoblastic lymphoma patient in the context of Li-Fraumeni syndrome. J. Immunother. Cancer 2020, 8, e000364.
- Keegan, T.H.M.; Bleyer, A.; Rosenberg, A.S.; Li, Q.; Goldfarb, M. Second primary malignant neoplasms and survival in adolescent and young adult cancer survivors. JAMA Oncol. 2017, 3, 1554–1557.
- Mullard, A. p53 programmes plough on. Nat. Rev. Drug Discov. 2020, 19, 497–500.
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 2006, 22, 741–753.
- Liu, J.; Guan, D.; Dong, M.; Yang, J.; Wei, H.; Liang, Q.; Song, L.; Xu, L.; Bai, J.; Liu, C.; et al. UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat. Cell Biol. 2020, 22, 1056–1063.
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577.
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299.
- Joseph, T.W.; Zaika, A.; Moll, U.M. Nuclear and cytoplasmic degradation of endogenous p53 and HDM2 occurs during down-regulation of the p53 response after multiple types of DNA damage. FASEB J. 2003, 17, 1622–1630.
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480.
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758.
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805.
- Kruse, J.-P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622.
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79.
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168.
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16.
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191.
- Giono, L.E.; Resnick-Silverman, L.; Carvajal, L.A.; St Clair, S.; Manfredi, J.J. Mdm2 promotes Cdc25C protein degradation and delays cell cycle progression through the G2/M phase. Oncogene 2017, 36, 6762–6773.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420.
- Raj, N.; Attardi, L.D. The transactivation domains of the p53 protein. Cold Spring Harb. Perspect. Med. 2017, 7, a026047.
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953.
- Böttger, A.; Böttger, V.; Garcia-Echeverria, C.; Chène, P.; Hochkeppel, H.K.; Sampson, W.; Ang, K.; Howard, S.F.; Picksley, S.M.; Lane, D.P. Molecular characterization of the hdm2-p53 interaction. J. Mol. Biol. 1997, 269, 744–756.
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482.
- Espinoza-Fonseca, L.M. Leucine-rich hydrophobic clusters promote folding of the N-terminus of the intrinsically disordered transactivation domain of p53. FEBS Lett. 2009, 583, 556–560.
- Dickinson, E.R.; Jurneczko, E.; Nicholson, J.; Hupp, T.R.; Zawacka-Pankau, J.; Selivanova, G.; Barran, P.E. The use of ion mobility mass spectrometry to probe modulation of the structure of p53 and of MDM2 by small molecule inhibitors. Front. Mol. Biosci. 2015, 2, 39.
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.G.C.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328.
- Rippin, T.M.; Freund, S.M.V.; Veprintsev, D.B.; Fersht, A.R. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol. 2002, 319, 351–358.
- Tanaka, T.; Watanabe, M.; Yamashita, K. Potential therapeutic targets of TP53 gene in the context of its classically canonical functions and its latest non-canonical functions in human cancer. Oncotarget 2018, 9, 16234–16247.
- Zawacka-Pankau, J.; Krachulec, J.; Grulkowski, I.; Bielawski, K.P.; Selivanova, G. The p53-mediated cytotoxicity of photodynamic therapy of cancer: Recent advances. Toxicol. Appl. Pharmacol. 2008, 232, 487–497.
- Melino, G.; Lu, X.; Gasco, M.; Crook, T.; Knight, R.A. Functional regulation of p73 and p63: Development and cancer. Trends Biochem. Sci. 2003, 28, 663–670.
- Ethayathulla, A.S.; Nguyen, H.T.; Viadiu, H. Crystal structures of the DNA-binding domain tetramer of the p53 tumor suppressor family member p73 bound to different full-site response elements. J. Biol. Chem. 2013, 288, 4744–4754.
- Wiederstein, M.; Sippl, M.J. TopMatch-web: Pairwise matching of large assemblies of protein and nucleic acid chains in 3D. Nucleic Acids Res. 2020, 48, W31–W35.
- Tan, Y.S.; Mhoumadi, Y.; Verma, C.S. Roles of computational modelling in understanding p53 structure, biology, and its therapeutic targeting. J. Mol. Cell Biol. 2019, 11, 306–316.
- Demir, Ö.; Ieong, P.U.; Amaro, R.E. Full-length p53 tetramer bound to DNA and its quaternary dynamics. Oncogene 2017, 36, 1451–1460.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555-572 .
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197.
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102.
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83.
- Barak, Y.; Gottlieb, E.; Juven-Gershon, T.; Oren, M. Regulation of mdm2 expression by p53: Alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994, 8, 1739–1749.
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602.
- Okamoto, K.; Tsunematsu, R.; Tahira, T.; Sonoda, K.; Asanoma, K.; Yagi, H.; Yoneda, T.; Hayashi, K.; Wake, N.; Kato, K. SNP55, a new functional polymorphism of MDM2-P2 promoter, contributes to allele-specific expression of MDM2 in endometrial cancers. BMC Med. Genet. 2015, 16, 67.
- Helwa, R.; Gansmo, L.B.; Romundstad, P.; Hveem, K.; Vatten, L.; Ryan, B.M.; Harris, C.C.; Lønning, P.E.; Knappskog, S. MDM2 promoter SNP55 (rs2870820) affects risk of colon cancer but not breast-, lung-, or prostate cancer. Sci. Rep. 2016, 6, 33153.
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014.
- Tao, W.; Levine, A.J. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of MDM2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941.
- Esteller, M.; Cordon-Cardo, C.; Corn, P.G.; Meltzer, S.J.; Pohar, K.S.; Watkins, D.N.; Capella, G.; Peinado, M.A.; Matias-Guiu, X.; Prat, J.; et al. p14ARF silencing by promoter hypermethylation mediates abnormal intracellular localization of MDM2. Cancer Res. 2001, 61, 2816–2821.
- Lindström, M.S.; Klangby, U.; Wiman, K.G. p14ARF homozygous deletion or MDM2 overexpression in Burkitt lymphoma lines carrying wild type p53. Oncogene 2001, 20, 2171–2177.
- Berggren de Verdier, P.J.; Kumar, R.; Adolfsson, J.; Larsson, P.; Norming, U.; Onelöv, E.; Wijkström, H.; Steineck, G.; Hemminki, K. Prognostic significance of homozygous deletions and multiple duplications at the CDKN2A (p16INK4a)/ARF (p14ARF) locus in urinary bladder cancer. Scand. J. Urol. Nephrol. 2006, 40, 363–369.
- Horn, H.F.; Vousden, K.H. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 2008, 27, 5774–5784.
- Zhou, X.; Hao, Q.; Zhang, Q.; Liao, J.M.; Ke, J.W.; Liao, P.; Cao, B.; Lu, H. Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ. 2015, 22, 755–766.
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288.
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439.
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388.
- Bykov, V.J.N.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 2016, 6, 21.
- Sallman, D.A. To target the untargetable: Elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1470–1472.
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848.
- Patton, J.T.; Mayo, L.D.; Singhi, A.D.; Gudkov, A.V.; Stark, G.R.; Jackson, M.W. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res. 2006, 66, 3169–3176.
- Joseph, T.L.; Madhumalar, A.; Brown, C.J.; Lane, D.P.; Verma, C.S. Differential binding of p53 and nutlin to MDM2 and MDMX: Computational studies. Cell Cycle 2010, 9, 1167–1181.
- Marine, J.C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934.
- Tournillon, A.S.; López, I.; Malbert-Colas, L.; Findakly, S.; Naski, N.; Olivares-Illana, V.; Karakostis, K.; Vojtesek, B.; Nylander, K.; Fåhraeus, R. p53 binds the mdmx mRNA and controls its translation. Oncogene 2017, 36, 723–730.
- Tolcher, A.W.; Karim, R.; Tang, Y.; Ji, J.; Wang, H.; Meng, L.; Kaiser, A.; Coe, J.; Liang, E.; Rosas, C.; et al. Abstract A086: Phase Ib study of a novel MDM2 inhibitor APG-115, in combination with pembrolizumab in patients with metastatic solid tumors in U.S. [abstract]. In: Proceedings of the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2019 Oct 26-30; Boston, MA. Philadelphia (PA): AACR (American Association for Cancer Research); Mol Cancer Ther 2019; 18(12 Suppl):Abstract nr A086.
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003.
- Jost, C.A.; Marin, M.C.; Kaelin, W.G. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997, 389, 191–194.
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819.
- Luh, L.M.; Kehrloesser, S.; Deutsch, G.B.; Gebel, J.; Coutandin, D.; Schäfer, B.; Agostini, M.; Melino, G.; Dötsch, V. Analysis of the oligomeric state and transactivation potential of TAp73α. Cell Death Differ. 2013, 20, 1008–1016.
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386.
- Ferraiuolo, M.; Di Agostino, S.; Blandino, G.; Strano, S. Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front. Oncol. 2016, 6, 77.
- Stantic, M.; Wolfsberger, J.; Sakil, H.A.M.; Wilhelm, M.T. ΔNp73 enhances HIF-1α protein stability through repression of the ECV complex. Oncogene 2018, 37, 3729–3739.
- Sakil, H.A.M.; Stantic, M.; Wolfsberger, J.; Brage, S.E.; Hansson, J.; Wilhelm, M.T. ΔNp73 regulates the expression of the multidrug-resistance genes ABCB1 and ABCB5 in breast cancer and melanoma cells—A short report. Cell. Oncol. 2017, 40, 631–638.
- Niemantsverdriet, M.; Nagle, P.; Chiu, R.K.; Langendijk, J.A.; Kampinga, H.H.; Coppes, R.P. ΔNp73 enhances promoter activity of TGF-β induced genes. PLoS ONE 2012, 7, e50815.
- Vella, V.; Puppin, C.; Damante, G.; Vigneri, R.; Sanfilippo, M.; Vigneri, P.; Tell, G.; Frasca, F. DeltaNp73alpha inhibits PTEN expression in thyroid cancer cells. Int. J. Cancer 2009, 124, 2539–2548.
- Zawacka-Pankau, J.; Kostecka, A.; Sznarkowska, A.; Hedström, E.; Kawiak, A. p73 tumor suppressor protein: A close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle 2010, 9, 720–728.
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137.
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: Regulators and effectors. Hum. Mutat. 2014, 35, 702–714.
- D’Alessandro, A.; Marrocco, C.; Rinalducci, S.; Peschiaroli, A.; Timperio, A.M.; Bongiorno-Borbone, L.; Finazzi Agrò, A.; Melino, G.; Zolla, L. Analysis of TAp73-dependent signaling via omics technologies. J. Proteome Res. 2013, 12, 4207–4220.
- Agostini, M.; Annicchiarico-Petruzzelli, M.; Melino, G.; Rufini, A. Metabolic pathways regulated by TAp73 in response to oxidative stress. Oncotarget 2016, 7, 29881–29900.
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3, e285.
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene 1999, 18, 2101–2106.
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y. MDM2 and MDMX bind and stabilize the p53-related protein p73. Curr. Biol. 1999, 9, 829–832.
- Zdzalik, M.; Pustelny, K.; Kedracka-Krok, S.; Huben, K.; Pecak, A.; Wladyka, B.; Jankowski, S.; Dubin, A.; Potempa, J.; Dubin, G. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010, 9, 4584–4591.
- Mavinahalli, J.N.; Madhumalar, A.; Beuerman, R.W.; Lane, D.P.; Verma, C. Differences in the transactivation domains of p53 family members: A computational study. BMC Genom. 2010, 11 (Suppl. 1), S5.
- Zeng, X.; Li, X.; Miller, A.; Yuan, Z.; Yuan, W.; Kwok, R.P.; Goodman, R.; Lu, H. The N-terminal domain of p73 interacts with the CH1 domain of p300/CREB binding protein and mediates transcriptional activation and apoptosis. Mol. Cell. Biol. 2000, 20, 1299–1310.
- Zuckerman, V.; Lenos, K.; Popowicz, G.M.; Silberman, I.; Grossman, T.; Marine, J.-C.; Holak, T.A.; Jochemsen, A.G.; Haupt, Y. c-Abl phosphorylates Hdmx and regulates its interaction with p53. J. Biol. Chem. 2009, 284, 4031–4039.
- Sionov, R.V.; Moallem, E.; Berger, M.; Kazaz, A.; Gerlitz, O.; Ben-Neriah, Y.; Oren, M.; Haupt, Y. c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J. Biol. Chem. 1999, 274, 8371–8374.
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 276, 15164–15173.
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007, 14, 743–751.
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.-C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848.
- Bálint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929.
- Wang, X.; Arooz, T.; Siu, W.Y.; Chiu, C.H.; Lau, A.; Yamashita, K.; Poon, R.Y. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001, 490, 202–208.
- Kubo, N.; Okoshi, R.; Nakashima, K.; Shimozato, O.; Nakagawara, A.; Ozaki, T. MDM2 promotes the proteasomal degradation of p73 through the interaction with Itch in HeLa cells. Biochem. Biophys. Res. Commun. 2010, 403, 405–411.
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492.
- Yoon, M.-K.; Kim, B.-Y.; Lee, J.-Y.; Ha, J.-H.; Kim, S.A.; Lee, D.-H.; Lee, M.-S.; Lee, M.-K.; Choi, J.S.; Cho, J.H.; et al. Cytoplasmic pro-apoptotic function of the tumor suppressor p73 is mediated through a modified mode of recognition of the anti-apoptotic regulator Bcl-XL. J. Biol. Chem. 2018, 293, 19546–19558.
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221.
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373.
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.-S.; et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008, 22, 2677–2691.
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015, 517, 626–630.
- Feeley, K.P.; Adams, C.M.; Mitra, R.; Eischen, C.M. Mdm2 Is Required for Survival and Growth of p53-Deficient Cancer Cells. Cancer Res. 2017, 77, 3823–3833.
- Amelio, I.; Inoue, S.; Markert, E.K.; Levine, A.J.; Knight, R.A.; Mak, T.W.; Melino, G. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1α degradation. Proc. Natl. Acad. Sci. USA 2015, 112, 226–231.
- Han, S.; Semba, S.; Abe, T.; Makino, N.; Furukawa, T.; Fukushige, S.; Takahashi, H.; Sakurada, A.; Sato, M.; Shiiba, K.; et al. Infrequent somatic mutations of the p73 gene in various human cancers. Eur. J. Surg. Oncol. 1999, 25, 194–198.
- Domínguez, G.; García, J.M.; Peña, C.; Silva, J.; García, V.; Martínez, L.; Maximiano, C.; Gómez, M.E.; Rivera, J.A.; García-Andrade, C.; et al. DeltaTAp73 upregulation correlates with poor prognosis in human tumors: Putative in vivo network involving p73 isoforms, p53, and E2F-1. J. Clin. Oncol. 2006, 24, 805–815.
- Hofstetter, G.; Berger, A.; Chamson, M.; Müller-Holzner, E.; Reimer, D.; Ulmer, H.; Uramoto, H.; Marth, C.; Zeimet, A.G.; Zeillinger, R.; et al. Clinical relevance of TAp73 and ΔNp73 protein expression in ovarian cancer: A series of 83 cases and review of the literature. Int. J. Gynecol. Pathol. 2011, 30, 527–531.
- Maas, A.-M.; Bretz, A.C.; Mack, E.; Stiewe, T. Targeting p73 in cancer. Cancer Lett. 2013, 332, 229–236.
- Hansen, T.M.; Rossi, M.; Roperch, J.P.; Ansell, K.; Simpson, K.; Taylor, D.; Mathon, N.; Knight, R.A.; Melino, G. Itch inhibition regulates chemosensitivity in vitro. Biochem. Biophys. Res. Commun. 2007, 361, 33–36.
- Lau, L.M.S.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene 2008, 27, 997–1003.
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307.
- Gong, J.G.; Costanzo, A.; Yang, H.Q.; Melino, G.; Kaelin, W.G.; Levrero, M.; Wang, J.Y. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809.
- Leong, C.-O.; Vidnovic, N.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Investig. 2007, 117, 1370–1380.
- Bunch, B.; Krishnan, N.; Greenspan, R.D.; Ramakrishnan, S.; Attwood, K.; Yan, L.; Qi, Q.; Wang, D.; Morrison, C.; Omilian, A.; et al. TAp73 expression and P1 promoter methylation, a potential marker for chemoresponsiveness to cisplatin therapy and survival in muscle-invasive bladder cancer (MIBC). Cell Cycle 2019, 18, 2055–2066.
- Spaety, M.-E.; Gries, A.; Badie, A.; Venkatasamy, A.; Romain, B.; Orvain, C.; Yanagihara, K.; Okamoto, K.; Jung, A.C.; Mellitzer, G.; et al. HDAC4 Levels Control Sensibility toward Cisplatin in Gastric Cancer via the p53-p73/BIK Pathway. Cancers 2019, 11, 1747.
- Kostecka, A.; Sznarkowska, A.; Meller, K.; Acedo, P.; Shi, Y.; Mohammad Sakil, H.A.; Kawiak, A.; Lion, M.; Królicka, A.; Wilhelm, M.; et al. JNK-NQO1 axis drives TAp73-mediated tumor suppression upon oxidative and proteasomal stress. Cell Death Dis. 2014, 5, e1484.
- Dabiri, Y.; Kalman, S.; Gürth, C.-M.; Kim, J.Y.; Mayer, V.; Cheng, X. The essential role of TAp73 in bortezomib-induced apoptosis in p53-deficient colorectal cancer cells. Sci. Rep. 2017, 7, 5423.



