 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicoletta Coccaro | + 3116 word(s) | 3116 | 2020-10-05 05:01:41 | | | |
| 2 | Bruce Ren | Meta information modification | 3116 | 2020-10-09 05:47:46 | | | | |
| 3 | Bruce Ren | Meta information modification | 3116 | 2020-10-26 08:46:45 | | |
Video Upload Options
The high mobility group AT-Hook (HMGA) proteins are a family of nonhistone chromatin remodeling proteins known as “architectural transcriptional factors”. By binding the minor groove of AT-rich DNA sequences, they interact with the transcription apparatus, altering the chromatin modeling and regulating gene expression by either enhancing or suppressing the binding of the more usual transcriptional activators and repressors, although they do not themselves have any transcriptional activity. Their involvement in both benign and malignant neoplasias is well-known and supported by a large volume of studies.
1. Introduction
The high mobility group AT-Hook (HMGA) proteins are a family of nonhistone chromatin remodeling proteins regulating gene expression, known as “architectural transcriptional factors” [1][2][3][4]. The gene family comprises the HMGA1 and HMGA2 genes; the former is located on chromosome 6 (6p21), is about 10 kb large, and consists of eight exons [5]; the latter is located on chromosome 12 (12q14-15), is ≥160 kb in size, and consists of five exons [6]. Alternative splicing of the HMGA1 gene produces three messenger RNA (mRNA) and three corresponding proteins, HMGA1a (formerly HMG-I), HMGA1b (formerly HMG-Y), and HMGA1c (formerly HMG-I/R) [7]. The HMGA1b isoform differs from the 1a isoform in that it lacks 11 amino acids; instead, the HMGA1c isoform is produced by alternative splicing of the HMGA1 gene using noncanonical acceptor and donor sites. As regards HMGA2, recently, two isoforms (HMGA2-L and HMGA2-S) have been described for which expression seems to be confined to the hematopoietic compartment and to depend on the stage of development of the hematopoietic stem cell (HSC) (see below) [8].
Sequence alignment showed that HMGA2 has a homology of about 55% with HMGA1 (A and B isoforms), including AT-hooks that are located in separate exons [6]. The AT-hook motif is positively charged and consists of a stretch of nine amino acids, containing the Arg-Gly-Arg-Pro repetition. This motif allows the protein to recognize and bind the AT-rich sequences in the minor groove of B-form DNA, rendering it able to recognize a structure rather than a specific sequence [9], so that some data report the ability of the HMGA1 protein to bind DNA even in regions without a rich AT content [10][11][12]. Once bound to DNA, the AT-hook undergoes a conformational change [13], and simultaneous binding of two or more AT-hooks to different binding sites leads to an increase in the strength of HMGA interactions with DNA [14].
By binding the minor groove of AT-rich DNA sequences, HMGA proteins interact with the transcription apparatus, altering the chromatin modeling and regulating gene expression by either enhancing or suppressing the binding of the more usual transcriptional activators and repressors, although they do not themselves have any transcriptional activity [15][16][17][18]. Their function is critical for the formation of higher-order nucleoprotein complexes (called enhanceosomes) in the promoter region, allowing the efficient transcriptional activation of a gene [19] thanks to their multiple surfaces capable of protein–protein interactions. Through such interactions, HMGA proteins may directly or indirectly control transcription, inducing conformational changes in chromatin or in the transcription factors themselves. In such ways, HMGA proteins are able to influence a wide variety of normal biological processes, such as cell growth, proliferation, differentiation, and death. They show an active role in the division cycle, and data indicate that both histone H1 and HMGA1 are necessary for chromatin condensation [20][21][22][23][24].
HMGA1 and 2 display different target genes; for example, the BRCA1 gene is negatively regulated by HMGA1 but not by HMGA2 [25]. Instead, HMGA2 has several other targets, including cycle regulators, one of which is the CDKN2A gene, encoding the two proteins p16INK4A and p14ARF. With the RNA-binding proteins LIN28A/B and the microRNA (miRNA) let-7b, HMGA2 and CDKN2A form an axis that controls stem cell aging in both normal and pathological conditions [22][23][24].
2. HMGA Proteins in Oncology
HMGA expression seems to be ubiquitous and high during embryogenesis and null or hardly detectable in adult differentiated tissues [26][27][28][29][30]. Several studies demonstrated the involvement of these proteins in embryonic development [31], adipocytes cell growth, and differentiation. Mice lacking the HMGA2 gene showed a pygmy phenotype with a drastic reduction of fat tissue [32]; instead, HMGA2 overexpression results in giant, obese mice [33]. On the contrary, HMGA1 inhibits adipocytes cell growth and triggers differentiation [34].
Overexpression of both genes is observed in most human malignant neoplasias, linking their alteration to carcinogenesis [35][36][37][38][39], while blocking their expression prevents thyroid cell transformation and triggers the death of malignant cells [40][41][42][43][44][45][46][47][48][49]. Various in vitro and in vivo studies have demonstrated the oncogenic potential of HMGA proteins; their overexpression transforms mouse and rat fibroblasts [50][51], and in transgenic mice, the development of natural killer T-cell lymphomas and pituitary adenomas has been observed [52][53][54][55]. There are high expression levels of HMGA transcripts in embryonic stem (ES) cells [56][57], HSCs [58][59][60], leukemic stem cells, and poorly differentiated or refractory tumors [61][62][63][64][65][66]. In cultured human lymphoid cells, the ectopic expression of HMGA1a triggers leukemic transformation [67] and, in transgenic mice, the development of an aggressive T-cell lymphoid neoplasia [68][69]. In particular, HMGA1 is overexpressed in several high-grade or refractory neoplasias, including hematologic malignancies and solid tumors [70]. Indeed, HMGA1 proteins are the most abundant nonhistone, chromatin-binding proteins in tumor cells .
romosomal duplications or translocations cause HMGA1 overexpression in rare cases; more frequently, the translocations involve HMGA2 (chromosomal region 12q14-15) in benign tumors, such as lipomas [71] and uterine fibroids [72][73][74]. With Fluorescent in Situ Hybridization (FISH) analysis, it has been observed that, in most cases, the breakpoint in the HMGA2 locus involves exons 1-2 or 1-3, leading to the loss of the C-terminal tail [75], separating the DNA-binding domains from the acid tails, and merging them with ectopic sequences. This seems to be sufficient for tumor development. Further studies revealed the presence of multiple binding sites for miRNA let-7 in the 3'-Untranslated Region (3’-UTR) of HMGA2 [76]. This evidence corroborates the observation that, in tumors with rearrangements destroying 3'-UTR of HMGA2, there were increased levels of the intact protein [77][78]; this suggests the presence of functional sites for the binding of miRNAs (such as let-7, mir-98, and miR-33) [79][65][66][78][79][65][66][80][81][82][83][84][85][86] that are important in regulating the gene expression.
A large volume of data demonstrates that miRNA dysregulation may intervene in tumor development [87][88]. Cancers exhibit distinctive miRNA expression signatures, raising the speculation that a dysregulation of specific pathways intervenes in tumorigenesis mechanisms in a specific tissue [89][90][91].
The repression of let-7 is necessary to enhance cell proliferation; in fact, let-7 expression is inversely related to the expression of HMGA2. This finding supports the hypothesis that let-7 can act as a repressor of HMGA2 [92].
3. HMGA Proteins in Hematopoiesis
In the hematologic field, HMGA proteins seem to play a pivotal role in both physiological and pathogenic conditions.
The loss of HMGA2 in HSCs does not allow a correct fetal hematopoietic process, causing slower self-renewal and proliferation rates, while it seems not to be indispensable in adult hematopoiesis [93]. Indeed, in 2013, in mouse models, Copley et al. demonstrated that the fine equilibrium between levels of the RNA-binding protein LIN28B and let-7 strikingly influence HSC developmentally timed changes, observing that HSC self-renewal potentials reduced when LIN28B decreased and let-7 increased [93]. As HMGA2 expression is high during embryogenesis, being particularly upregulated in fetal HSCs and gradually decreasing during the transition to adult hematopoiesis, experiments in transplanted irradiated hosts to define the role of HMGA2 and LIN28 in HSC functions showed that both LIN28- and HMGA2-induced overexpression increased the self-renewal activity of adult HSCs. However, HMGA2 overexpression in adult HSCs did not seem sufficient to mimic all the effects of elevated LIN28B that triggers a fetal lymphoid differentiation program. Accordingly, HMGA2 seems to be a downstream modulator of the HSC self-renewal ability; its function is regulated by LIN28B, that acts as a master regulator of developmentally timed changes of HSC [93]. However, this regulatory mechanism seems to be even finer tuned. In a recent work, Cesana et al. employed high-throughput genomic approaches to profile miRNAs, long intergenic non coding RNAs (lincRNAs), and mRNAs in HSCs during development in order to characterize transcriptional and posttranscriptional changes. These analyses highlighted distinct alternative splicing patterns of HSCs in various key hematopoietic regulators, one of which is HMGA2, for which an alternative isoform, escaping miRNA-mediated control, was identified. It seems that the splicing kinase CLK3 conserves HMGA2 function in response to an increase of let-7 miRNA levels by regulating HMGA2 splicing. These data delineate a CLK3/HMGA2 functional axis regulating HSC functions during development. In humans, HMGA2 presents alternative splicing isoforms, HMGA2-L, which is sensitive to degradation by let-7, being the dominant isoform in fetal HSCs, whereas newborn HSCs express the HMGA2-S isoform, that is resistant to degradation by let-7 (Figure 1). In support of the crucial role of HMGA2 in regulating HSCs properties, it has been found in gene therapy experiments that a deliberate overexpression of HMGA2, in parallel with that of the transgene of interest, might be needed to obtain the therapeutic effect [94][95], particularly in cases where a large fraction of corrected HSCs must be obtained to gain the benefit. In the case reported in 2010 by Cavazzana et al. of an adult patient with severe βE/β0-thalassaemia treated with gene therapy, the presence of a dominant, myeloid-biased cell clone was demonstrated, in which the integrated vector transferring the β-globin gene caused the transcriptional activation of HMGA2 in erythroid cells and further increased the expression of a truncated HMGA2 mRNA insensitive to degradation by let-7 miRNAs [94].
There is evidence in mice that Hmga2 is also a regulator of gamma-globin mRNA, and that it moderately increased HbF levels in human adult erythroblasts in vitro [96]. Targeted inhibition of let-7 is sufficient for specific developmental changes to occur in gamma-globin transcription and HbF levels through increased HMGA2 levels [97].
As regards the LIN28B-let7-HMGA2 axis, it is also involved in the megakaryocyte and platelet lineage. LIN28B expression in fetal myelo-erythroid progenitors suggests that it could have a role in the prenatal platelet-forming lineages; indeed, megakaryocytes derived from human ES cells express higher levels of LIN28B as compared to adult controls [98]. This implies that LIN28B may also have an active function in megakaryocyte development and thus platelet function [99].
In vitro experiments on ES cells lacking one or both HMGA1 alleles showed that these cells showed a lesser differentiation in T-cell precursors than Wild Type (WT) ES cells and preferentially in B cells, maybe because of an increased interleukin 6 and decreased interleukin 2 expression [100]. Furthermore, there was evidence of an aberrant hemopoietic differentiation, with a reduction of the monocyte/macrophage population and an increase in megakaryocyte precursors, erythropoiesis, and globin gene expression. The restoration of HMGA1 expression reestablished the WT phenotype. These studies highlighted that HMGA1 proteins directly control the transcription of GATA-1, a key transcription factor which regulates red blood cell differentiation, that is overexpressed in HMGA1−/− ES cells. This could offer an explanation, at least in part, of the alteration of the lineages of megakaryocytes/erythrocytes [100]. These observations add weight to those of Pierantoni et al., who observed that the induction of HMGA1 protein synthesis appears to play a role in the differentiation of megakaryocytes while its inhibition may be necessary in the differentiation of erythrocytes or macrophages [101].
Regarding lymphoid compartment, animal experiments showed HMGA2 involvement in mouse thymopoiesis, as it is highly expressed in fetal and neonatal early T cell progenitors (ETP), the most intrathymic precursors, while it results almost undetectable 5 weeks after birth [102]. In the same way, the number of thymic cells increases during the first two weeks after birth, stabilizes, and then declines by seven weeks of age. In fact, HMGA2-deficient mice showed deficient ETPs after birth, and also the total thymocyte number was fewer as compared to WT. Bearing in mind that, also in human fetal thymic progenitors, HMGA2 expression is high and decreases during growth, it is possible that a similar mechanism takes place [102].
As regards HMGA1, studies using HMGA1-green fluorescent protein fusion explored HMGA1 expression in undifferentiated and differentiated populations of hematopoietic cells, demonstrating that HMGA1 is highly expressed in CD4/CD8-double negative (DN) cells and transiently downregulated in CD4⁄CD8-double positive (DP) cells during early T cell development in the thymus [103]. Indeed, HMGA1 binds directly to cis-regulatory elements in the CD4 ⁄ CD8 loci in DN cells but not in DP cells. Moreover, in DN leukemic cells, CD4⁄CD8 expression is induced by the inhibition of HMGA1, and T leukemic cells lacking HMGA1 showed a markedly decreased proliferation [103].
HMGA1 indirectly controls µ enhancer activity during B lymphocyte development, acting on the combinatorial activity of transcription factors [104][105][106][107]. Various studies revealed that HMGA1 interacts with both PU.1 and ETS proto-oncogene 1 transcription factor 1 (Ets-1) in solution [108][109]; MGA1 interaction with PU.1 likely induces a change in PU.1 structure, increasing PU.1/µ enhancer binding. This augmented binding renders HMGA1 able to boost PU.1/Ets-1 functional synergy on the µ enhancer [110].
In view of the role of HMGA proteins in driving both benign and malignant tumors and the evidence regarding their main functions in various hematopoietic physiological mechanisms, various research studies have explored their involvement in hematological malignancies.
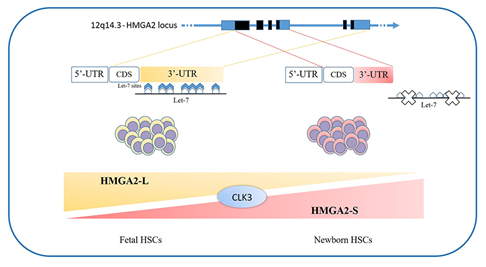
Figure 1. HMGA2 development-specific isoforms: HSCs feature distinct alternative splicing patterns in various key hematopoietic regulators, one of which is HMGA2. In humans, two alternative splicing isoforms have recently been discovered: the HMGA2-L isoform, which is sensitive to degradation by let-7 and is dominant in fetal HSCs, and the HMGA2-S isoform, that is resistant to degradation by let-7 and is prevalent in newborn HSCs. The two isoforms reveal alternative 3’UTR usage; they share the first three exons and differ in their terminal exon usage (including C-terminal domains and 3’UTRs). The HMGA2-S 3’UTR is one third the length of the HMGA2-L 3’UTR and lacks the seven let-7 sites as well as the other most conserved miRNA sites. The balance between alternative isoforms and the escape from miRNA-mediated control seems to depend on the activity of the splicing kinase CLK3, which promotes exon skipping and thus influences the HMGA2 splicing pattern. HSCs - hematopoietic stem cells; 3’UTR - 3'-Untranslated Region.[111]
4. HMGA Proteins as Targetable Markers
In view of their role in carcinogenesis, HMGA proteins could be suitable candidates for molecular therapy against HMGA-overexpressing cancers. As they bind AT-specific minor grooves of DNA, their binding could be inhibited by other DNA minor groove binders/ligands, such as distamycin and netropsin [112], that have been studied as potential new therapies for several types of human cancer [113].
The search for HMGA1 inhibitors is ongoing. Despite toxicity issues that still need to be addressed, promising preliminary data have been obtained for flavopiridol and FR900482 [114].
A series of studies explored the use of STAT3 as a targetable marker in aggressive lymphoid malignancies overexpressing HMGA1. Nanoparticle delivery of STAT3 with G-quartet oligodeoxynucleotides that specifically inhibit STAT3 binding to DNA has been shown to have antileukemia effects in an HMGA1 transgenic model of aggressive T-ALL [115]. Indeed, BP-1-102, a salicylic acid-based inhibitor, was tested in preclinical models of ALL and Burkitts lymphoma with STAT3 activation [116], demonstrating that BP-1-102 inhibits STAT3 phosphorylation at tyrosine 705, preventing STAT3 dimerization, DNA binding, and the activation of downstream genes. It also decreases nuclear and cytoplasmic phosphorylated STAT3. Further treatment of cultured cells derived from B-lymphoid tumors previously shown to express high levels of HMGA1 and STAT3, including B-ALL, Burkitts leukemia, and T-ALL cells, showed a decreased proliferation [117]. However, there was no antitumor efficacy in murine xenograft models; the reason for this is still uncertain and may lie either in the limited exposure to the drug or in the induction of oncogenic pathways alternative to the one inhibited by BP-1-102[117]. These experiments also helped to show that BP-1-102 treatment represses HMGA1, highlighting that not only does HMGA1 induce STAT3 expression but even STAT3 feeds forward to upregulate HMGA1, leading to an enhanced expression of both genes during tumor progression. In fact, a putative STAT3 consensus DNA binding site (TTN5AA) was found in the HMGA1 promoter region; it is conserved in humans and mice and could mediate STAT3 dimer binding and transactivation. In conjunction with direct transactivation, STAT3 could induce downstream factors that upregulate HMGA1 expression [117].
Furthermore, Bortezomib (BTZ) also seems to influence HMGA1 levels. This is a proteasome inhibitor used to suppress Diffuse large B-cell lymphoma (DLBCL) progression [118][119][120]. DLBCL is the most frequent non-Hodgkin lymphoma, showing variable clinical presentations as well as variable therapy responses [121][122][123]. In vitro studies on DLBCL CRL-2630 cells showed that BTZ treatment significantly inhibited the proliferation of DLBCL CRL-2630 cells [124]. After studies showing that exposure to BTZ induced an upregulation of miRNA 198 (miR-198) while depletion of the miR significantly reversed the inhibitory effect of BTZ on cell proliferation [125], further studies on miR-198 revealed the 3’-UTR of HMGA1 as a downstream target of miR-198, suppressing the expression of HMGA1 in DLBCL cells. This evidence links HMGA1 to BTZ treatment that decreased the level of HMAG1 and inhibited the migration of DLBCL cells [126][127][128].
Indeed, a recent screening of about 36,000 compounds indicated a class of phosphodiesterase inhibitors that help to suppress let-7 targets [129]. These potential drugs augment cAMP levels and elevate let-7 levels, inhibiting let-7 target genes such as HMGA2 and MYC and decreasing growth in multiple cancer cell lines [129].
In the treatment of CML patients, the introduction of specific tyrosine kinase inhibitors (TKI) has promoted great progress in patient management [130][131]. Several TKIs are currently enrolled in common practice (such as imatinib, nilotinib, and dasatinib), the use of which is strictly related to adverse events and to the observation of resistance phenomena developing during therapy [132][133]. These issues increase the need to devise alternative therapies; in recent years, studies on the use of epigenetic therapy associated with other therapies have seemed to offer valid therapeutic tools[134]. In this regard, the Vitkeviciene team investigated the potential role of two epigenetic modulators, EGCG (epigallocatechin-3-gallate) and BIX-01294 (1-benzylpiperidin-4-yl)-6,7-dimethoxy-2-(4-methyl-1,4-diazepan-1-yl) quinazolin-4-amine) in two AML and CML cell lines [135].
EGCG is a naturally occurring catechin in green tea. This molecule is known to have many functions, including antibacterial, antioxidant, and anti-inflammatory actions, but it also seems to have a potential antineoplastic role, inhibiting proliferation and inducing apoptosis in different tumors [136][137]. BIX-01294 is a synthetic molecule that allows the specific inhibition of EHMT2/G9a histone methyltransferase. EHMT2/G9a is an important protagonist of gene silencing mechanisms[138] [139]. A study in vitro showed that EGCG and BIX-01294, acting as epigenetic modulators, cause cell cycle arrest in the G0/G1 phase; the lines treated with EGCG also showed an increased level of ATM, HMGA2, phosphorylation of ATM, and Senescence-associated (SA)-β-galactosidase staining, for which concerted action favors the cellular senescence phenomenon [135]. Although apoptosis as compared to the induction of senescence is believed to be more effective in the treatment of cancer, the use of substances capable of inducing senescence offers a great advantage in terms of therapy planning [140].
References
- Reeves, R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene 2001, 277, 63–81.
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910.
- Resar, L.M.S. The high mobility group A1 gene: transforming inflammatory signals into cancer? Cancer Res. 2010, 70, 436–9.
- Shah, S.N.; Resar, L.M.S. High mobility group A1 and cancer: potential biomarker and therapeutic target. Histol. Histopathol. 2012, 27, 567–79.
- Friedmann, M.; Holth, L.T.; Zoghbi, H.Y.; Reeves, R. Organization, inducible-expression and chromosome localization of the human HMG-I(Y) nonhistone protein gene. Nucleic Acids Res. 1993, 21, 4259–67.
- Cleynen, I.; Van de Ven, W.J.M. The HMGA proteins: a myriad of functions (Review). Int. J. Oncol. 2008, 32, 289–305.
- Nagpal, S.; Ghosn, C.; DiSepio, D.; Molina, Y.; Sutter, M.; Klein, E.S.; Chandraratna, R.A. Retinoid-dependent recruitment of a histone H1 displacement activity by retinoic acid receptor. J. Biol. Chem. 1999, 274, 22563–8.
- Cesana, M.; Guo, M.H.; Cacchiarelli, D.; Wahlster, L.; Barragan, J.; Doulatov, S.; Vo, L.T.; Salvatori, B.; Trapnell, C.; Clement, K.; et al. A CLK3-HMGA2 Alternative Splicing Axis Impacts Human Hematopoietic Stem Cell Molecular Identity throughout Development. Cell Stem Cell 2018, 22, 575-588.e7.
- Zhang, S.; Mo, Q.; Wang, X. Oncological role of HMGA2 (Review). Int. J. Oncol. 2019, 55, 775–778.
- Reeves, R.; Wolffe, A.P. Substrate structure influences binding of the non-histone protein HMG-I(Y) to free and nucleosomal DNA. Biochemistry 1996, 35, 5063–5074.
- Nissen, M.S.; Reeves, R. Changes in superhelicity are introduced into closed circular DNA by binding of high mobility group protein I/Y. J. Biol. Chem. 1995, 270, 4355–4360.
- Hill, D.A.; Pedulla, M.L.; Reeves, R. Directional binding of HMG-I(Y) on four-way junction DNA and the molecular basis for competitive binding with HMG-1 and histone H1. Nucleic Acids Res. 1999, 27, 2135–44.
- Reeves, R.; Beckerbauer, L. HMGI/Y proteins: Flexible regulators of transcription and chromatin structure. Biochim. Biophys. Acta - Gene Struct. Expr. 2001, 1519, 13–29.
- Frank, O.; Schwanbeck, R.; Wiśniewski, J.R. Protein footprinting reveals specific binding modes of a high mobility group protein I to DNAs of different conformation. J. Biol. Chem. 1998, 273, 20015–20020.
- Thanos, D.; Maniatis, T. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 1995, 83, 1091–1100.
- Thanos, D.; Du, W.; Maniatis, T. The high mobility group protein HMG I(Y) is an essential structural component of a virus-inducible enhancer complex. In Proceedings of the Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press, 1993, 58, 73–81.
- Thanos, D.; Maniatis, T. The High Mobility Group protein HMG I(Y) is required for NF-κB-dependent virus induction of the human IFN-β gene. Cell 1992, 71, 777–789.
- Wolffe, A.P. Architectural transcription factors. Science 1994, 264, 1100–1.
- Carey, M. The enhanceosome and transcriptional synergy. Cell 1998, 92, 5–8.
- Radic, M.Z.; Saghbini, M.; Elton, T.S.; Reeves, R.; Hamkalo, B.A. Hoechst 33258, distamycin A, and high mobility group protein I (HMG-I) compete for binding to mouse satellite DNA. Chromosoma 1992, 101, 602–608.
- Käs, E.; Poljak, L.; Adachi, Y.; Laemmli, U.K. A model for chromatin opening: stimulation of topoisomerase II and restriction enzyme cleavage of chromatin by distamycin. EMBO J. 1993, 12, 115–126.
- Baldassarre, G.; Battista, S.; Belletti, B.; Thakur, S.; Pentimalli, F.; Trapasso, F.; Fedele, M.; Pierantoni, G.; Croce, C.M.; Fusco, A. Negative Regulation of BRCA1 Gene Expression by HMGA1 Proteins Accounts for the Reduced BRCA1 Protein Levels in Sporadic Breast Carcinoma. Mol. Cell. Biol. 2003, 23, 2225–2238.
- Hammond, S.M.; Sharpless, N.E. HMGA2, MicroRNAs, and Stem Cell Aging. Cell 2008, 135, 1013–1016.
- Nishino, J.; Kim, I.; Chada, K.; Morrison, S.J. Hmga2 promotes neural stem cell self-renewal in young but not olNishino, J., Kim, I., Chada, K., & Morrison, S. J. (2008). Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 2008, 135, 227–39.
- Tzatsos, A.; Bardeesy, N. Ink4a/Arf regulation by let-7b and Hmga2: a genetic pathway governing stem cell aging. Cell Stem Cell 2008, 3, 469–70.
- Zhou, X.; Benson, K.F.; Przybysz, K.; Liu, J.; Hou, Y.; Cherath, L.; Chada, K. Genomic structure and expression of the murine Hmgi-c gene. Nucleic Acids Res. 1996, 24, 4071–7.
- Rogalla, P.; Drechsler, K.; Frey, G.; Hennig, Y.; Helmke, B.; Bonk, U.; Bullerdiek, J. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am. J. Pathol. 1996, 149, 775–9.
- Rommel, B.; Rogalla, P.; Jox, A.; Kalle, C. V; Kazmierczak, B.; Wolf, J.; Bullerdiek, J. HMGI-C, a member of the high mobility group family of proteins, is expressed in hematopoietic stem cells and in leukemic cells. Leuk. Lymphoma 1997, 26, 603–7.
- Gattas, G.J.; Quade, B.J.; Nowak, R.A.; Morton, C.C. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes. Chromosomes Cancer 1999, 25, 316–22.
- Somervaille, T.C.P.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–40.
- Chiappetta, G.; Avantaggiato, V.; Visconti, R.; Fedele, M.; Battista, S.; Trapasso, F.; Merciai, B.M.; Fidanza, V.; Giancotti, V.; Santoro, M.; et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996, 13, 2439–2446.
- Zhou, X.; Benson, K.F.; Ashar, H.R.; Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995, 376, 771–4.
- Battista, S.; Fidanza, V.; Fedele, M.; Klein-Szanto, A.J.; Outwater, E.; Brunner, H.; Santoro, M.; Croce, C.M.; Fusco, A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999, 59, 4793–7.
- Melillo, R.M.; Pierantoni, G.M.; Scala, S.; Battista, S.; Fedele, M.; Stella, A.; De Biasio, M.C.; Chiappetta, G.; Fidanza, V.; Condorelli, G.; et al. Critical Role of the HMGI(Y) Proteins in Adipocytic Cell Growth and Differentiation. Mol. Cell. Biol. 2001, 21, 2485–2495.
- Tallini, G.; Dal Cin, P. HMGI(Y) and HMGI-C dysregulation: a common occurrence in human tumors. Adv. Anat. Pathol. 1999, 6, 237–46.
- Harada-Shirado, K.; Ikeda, K.; Ogawa, K.; Ohkawara, H.; Kimura, H.; Kai, T.; Noji, H.; Morishita, S.; Komatsu, N.; Takeishi, Y. Dysregulation of the MIRLET7/HMGA2 axis with methylation of the CDKN2A promoter in myeloproliferative neoplasms. Br. J. Haematol. 2015, 168, 338–49.
- Huang, M.L.; Chen, C.C.; Chang, L.C. Gene expressions of HMGI-C and HMGI(Y) are associated with stage and metastasis in colorectal cancer. Int. J. Colorectal Dis. 2009, 24, 1281–1286.
- Wang, X.; Liu, X.; Li, A.Y.-J.; Chen, L.; Lai, L.; Lin, H.H.; Hu, S.; Yao, L.; Peng, J.; Loera, S.; et al. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin. Cancer Res. 2011, 17, 2570–80.
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front. Med. 2015, 2, 15.
- Berlingieri, M.T.; Pierantoni, G.M.; Giancotti, V.; Santoro, M.; Fusco, A. Thyroid cell transformation requires the expression of the HMGA1 proteins. Oncogene 2002, 21, 2971–80.
- Scala, S.; Portella, G.; Fedele, M.; Chiappetta, G.; Fusco, A. Adenovirus-mediated suppression of HMGI(Y) protein synthesis as potential therapy of human malignant neoplasias. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 4256–61.
- Berlingieri, M.T.; Manfioletti, G.; Santoro, M.; Bandiera, A.; Visconti, R.; Giancotti, V.; Fusco, A. Inhibition of HMGI-C protein synthesis suppresses retrovirally induced neoplastic transformation of rat thyroid cells. Mol. Cell. Biol. 1995, 15, 1545–1553.
- Fedele, M.; Berlingieri, M.T.; Scala, S.; Chiariotti, L.; Viglietto, G.; Rippel, V.; Bullerdiek, J.; Santoro, M.; Fusco, A. Truncated and chimeric HMGI-C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene 1998, 17, 413–8.
- Wood, L.J.; Maher, J.F.; Bunton, T.E.; Resar, L.M. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000, 60, 4256–61.
- Baldassarre, G.; Fedele, M.; Battista, S.; Vecchione, A.; Klein-Szanto, A.J.P.; Santoro, M.; Waldmann, T.A.; Azimi, N.; Croce, C.M.; Fusco, A. Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI-C gene by the chronic stimulation of the IL-2 and IL-15 pathway. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7970–7975.
- Xu, Y.; Sumter, T.F.; Bhattacharya, R.; Tesfaye, A.; Fuchs, E.J.; Wood, L.J.; Huso, D.L.; Resar, L.M.S. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004, 64, 3371–5.
- Fedele, M.; Pentimalli, F.; Baldassarre, G.; Battista, S.; Klein-Szanto, A.J.P.; Kenyon, L.; Visone, R.; De Martino, I.; Ciarmiello, A.; Arra, C.; et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005, 24, 3427–35.
- Fedele, M.; Battista, S.; Kenyon, L.; Baldassarre, G.; Fidanza, V.; Klein-Szanto, A.J.P.; Parlow, A.F.; Visone, R.; Pierantoni, G.M.; Outwater, E.; et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene 2002, 21, 3190–3198.
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507.
- Chou, B.-K.; Mali, P.; Huang, X.; Ye, Z.; Dowey, S.N.; Resar, L.M.; Zou, C.; Zhang, Y.A.; Tong, J.; Cheng, L. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011, 21, 518–29.
- Karp, J.E.; Smith, B.D.; Resar, L.S.; Greer, J.M.; Blackford, A.; Zhao, M.; Moton-Nelson, D.; Alino, K.; Levis, M.J.; Gore, S.D.; et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood 2011, 117, 3302–10.
- Nelson, D.M.; Joseph, B.; Hillion, J.; Segal, J.; Karp, J.E.; Resar, L.M.S. Flavopiridol induces BCL-2 expression and represses oncogenic transcription factors in leukemic blasts from adults with refractory acute myeloid leukemia. Leuk. Lymphoma 2011, 52, 1999–2006.
- Zhou, G.; Chen, J.; Lee, S.; Clark, T.; Rowley, J.D.; Wang, S.M. The pattern of gene expression in human CD34(+) stem/progenitor cells. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13966–71.
- Dolde, C.E.; Mukherjee, M.; Cho, C.; Resar, L.M.S. HMG-I/Y in human breast cancer cell lines. Breast Cancer Res. Treat. 2002, 71, 181–91.
- Tesfaye, A.; Di Cello, F.; Hillion, J.; Ronnett, B.M.; Elbahloul, O.; Ashfaq, R.; Dhara, S.; Prochownik, E.; Tworkoski, K.; Reeves, R.; et al. The high-mobility group A1 gene up-regulates cyclooxygenase 2 expression in uterine tumorigenesis. Cancer Res. 2007, 67, 3998–4004.
- Di Cello, F.; Hillion, J.; Hristov, A.; Wood, L.J.; Mukherjee, M.; Schuldenfrei, A.; Kowalski, J.; Bhattacharya, R.; Ashfaq, R.; Resar, L.M.S. HMGA2 participates in transformation in human lung cancer. Mol. Cancer Res. 2008, 6, 743–50.
- Di Cello, F.; Hillion, J.; Kowalski, J.; Ronnett, B.M.; Aderinto, A.; Huso, D.L.; Resar, L.M.S. Cyclooxygenase inhibitors block uterine tumorigenesis in HMGA1a transgenic mice and human xenografts. Mol. Cancer Ther. 2008, 7, 2090–5.
- Hillion, J.; Dhara, S.; Sumter, T.F.; Mukherjee, M.; Di Cello, F.; Belton, A.; Turkson, J.; Jaganathan, S.; Cheng, L.; Ye, Z.; et al. The high-mobility group A1a/signal transducer and activator of transcription-3 axis: an achilles heel for hematopoietic malignancies? Cancer Res. 2008, 68, 10121–7.
- Hristov, A.C.; Cope, L.; Di Cello, F.; Reyes, M.D.; Singh, M.; Hillion, J.A.; Belton, A.; Joseph, B.; Schuldenfrei, A.; Iacobuzio-Donahue, C.A.; et al. HMGA1 correlates with advanced tumor grade and decreased survival in pancreatic ductal adenocarcinoma. Mod. Pathol. 2010, 23, 98–104.
- Belton, A.; Gabrovsky, A.; Bae, Y.K.; Reeves, R.; Iacobuzio-Donahue, C.; Huso, D.L.; Resar, L.M.S. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS One 2012, 7, e30034.
- Pomeroy, S.L.; Tamayo, P.; Gaasenbeek, M.; Sturla, L.M.; Angelo, M.; McLaughlin, M.E.; Kim, J.Y.H.; Goumnerova, L.C.; Black, P.M.; Lau, C.; et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002, 415, 436–442.
- Petit, M.M.R.; Mols, R.; Schoenmakers, E.F.P.M.; Mandahl, N.; Van De Ven, W.J.M. LPP, the preferred fusion partner gene of HMGIC in lipomas, is a novel member of the LIM protein gene family. Genomics 1996, 36, 118–129.
- Schoenmakers, E.F.; Huysmans, C.; Van de Ven, W.J. Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) uterine leiomyomas. Cancer Res. 1999, 59, 19–23.
- Schoenmakers, E.F.; Wanschura, S.; Mols, R.; Bullerdiek, J.; Van den Berghe, H.; Van de Ven, W.J. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat. Genet. 1995, 10, 436–44.
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science (80-. ). 2007.
- Wang, T.; Zhang, X.; Obijuru, L.; Laser, J.; Aris, V.; Lee, P.; Mittal, K.; Soteropoulos, P.; Wei, J.-J. A micro-RNA signature associated with race, tumor size, and target gene activity in human uterine leiomyomas. Genes. Chromosomes Cancer 2007, 46, 336–47.
- Wood, L.J.; Mukherjee, M.; Dolde, C.E.; Xu, Y.; Maher, J.F.; Bunton, T.E.; Williams, J.B.; Resar, L.M. HMG-I/Y, a new c-Myc target gene and potential oncogene. Mol. Cell. Biol. 2000, 20, 5490–502.
- Schuldenfrei, A.; Belton, A.; Kowalski, J.; Talbot, C.C.; Di Cello, F.; Poh, W.; Tsai, H.-L.; Shah, S.N.; Huso, T.H.; Huso, D.L.; et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics 2011, 12, 549.
- Efanov, A.; Zanesi, N.; Coppola, V.; Nuovo, G.; Bolon, B.; Wernicle-Jameson, D.; Lagana, A.; Hansjuerg, A.; Pichiorri, F.; Croce, C.M. Human HMGA2 protein overexpressed in mice induces precursor T-cell lymphoblastic leukemia. Blood Cancer J. 2014, 4, e227.
- Kazmierczak, B.; Meyer-Bolte, K.; Tran, K.H.; Wöckel, W.; Breightman, I.; Rosigkeit, J.; Bartnitzke, S.; Bullerdiek, J. A high frequency of tumors with rearrangements of genes of the HMGI(Y) family in a series of 191 pulmonary chondroid hamartomas. Genes. Chromosomes Cancer 1999, 26, 125–33.
- Odero, M.D.; Grand, F.H.; Iqbal, S.; Ross, F.; Roman, J.P.; Vizmanos, J.L.; Andrieux, J.; Laï, J.L.; Calasanz, M.J.; Cross, N.C.P. Disruption and aberrant expression of HMGA2 as a consequence of diverse chromosomal translocations in myeloid malignancies. Leukemia 2005, 19, 245–252.
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89.
- Geurts, J.M.W.; Schoenmakers, E.F.P.M.; Van De Ven, W.J.M. Molecular characterization of a complex chromosomal rearrangement in a pleomorphic salivary gland adenoma involving the 3’-UTR of HMGIC. Cancer Genet. Cytogenet. 1997, 95, 198–205.
- Inoue, N.; Izui-Sarumaru, T.; Murakami, Y.; Endo, Y.; Nishimura, J.I.; Kurokawa, K.; Kuwayama, M.; Shime, H.; Machii, T.; Kanakura, Y.; et al. Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood 2006, 108, 4232–4236.
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene Email alerting service The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 1025–1030.
- Hebert, C.; Norris, K.; Scheper, M.A.; Nikitakis, N.; Sauk, J.J. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 2007, 6.
- Baba, O.; Horie, T.; Nakao, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; Kuwabara, Y.; Nishiga, M.; Nishino, T.; Ide, Y.; et al. MicroRNA 33 Regulates the Population of Peripheral Inflammatory Ly6C high Monocytes through Dual Pathways . Mol. Cell. Biol. 2018, 38.
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquienelll, A.E.; Bettlnger, J.C.; Rougvle, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906.
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647.
- Grosveld, F.; van Assendelft, G.B.; Greaves, D.R.; Kollias, G. Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell 1987, 51, 975–985.
- Kottickal, L. V; Sarada, B.; Ashar, H.; Chada, K.; Nagarajan, L. Preferential expression of HMGI-C isoforms lacking the acidic carboxy terminal in human leukemia. Biochem. Biophys. Res. Commun. 1998, 242, 452–6.
- Santulli, B.; Kazmierczak, B.; Napolitano, R.; Caliendo, I.; Chiappetta, G.; Rippe, V.; Bullerdiek, J.; Fusco, A. A 12q13 translocation involving the HMGI-C gene in Richter transformation of a chronic lymphocytic leukemia. Cancer Genet. Cytogenet. 2000, 119, 70–73.
- Cirera-Salinas, D.; Pauta, M.; Allen, R.M.; Salerno, A.G.; Ramírez, C.M.; Chamorro-Jorganes, A.; Wanschel, A.C.; Lasuncion, M.A.; Morales-Ruiz, M.; Suarez, Y.; et al. Mir-33 regulates cell proliferation and cell cycle progression. Cell Cycle 2012, 11, 922–33.
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928.
- Rice, S.J.; Lai, S.-C.; Wood, L.W.; Helsley, K.R.; Runkle, E.A.; Winslow, M.M.; Mu, D. MicroRNA-33a mediates the regulation of high mobility group AT-hook 2 gene (HMGA2) by thyroid transcription factor 1 (TTF-1/NKX2-1). J. Biol. Chem. 2013, 288, 16348–60.
- Rice, S.J.; Lai, S.-C.; Wood, L.W.; Helsley, K.R.; Runkle, E.A.; Winslow, M.M.; Mu, D. MicroRNA-33a mediates the regulation of high mobility group AT-hook 2 gene (HMGA2) by thyroid transcription factor 1 (TTF-1/NKX2-1). J. Biol. Chem. 2013, 288, 16348–60.
- Wiemer, E.A.C. The role of microRNAs in cancer: No small matter. Eur. J. Cancer 2007, 43, 1529–1544.
- Godley, L.A. HMGA2 levels in CML: Reflective of miRNA gene regulation in a hematopoietic tumor? Leuk. Lymphoma 2007, 48, 1898–1899.
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838.
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677.
- Sgarra, R.; Pegoraro, S.; D’Angelo, D.; Ros, G.; Zanin, R.; Sgubin, M.; Petrosino, S.; Battista, S.; Manfioletti, A.G. High Mobility Group A (HMGA): Chromatin Nodes Controlled by a Knotty miRNA Network. Int. J. Mol. Sci. 2020, 21.
- Boyerinas, B.; Park, S.-M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008, 68, 2587–91.
- Copley, M.R.; Babovic, S.; Benz, C.; Knapp, D.J.H.F.; Beer, P.A.; Kent, D.G.; Wohrer, S.; Treloar, D.Q.; Day, C.; Rowe, K.; et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 2013, 15, 916–925.
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322.
- Wang, G.P.; Berry, C.C.; Malani, N.; Leboulch, P.; Fischer, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Bushman, F.D. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood 2010, 115, 4356–66.
- de Vasconcellos, J.F.; Lee, Y.T.; Byrnes, C.; Tumburu, L.; Rabel, A.; Miller, J.L. HMGA2 Moderately Increases Fetal Hemoglobin Expression in Human Adult Erythroblasts. PLoS One 2016, 11, e0166928.
- de Vasconcellos, J.F.; Byrnes, C.; Lee, Y.T.; Allwardt, J.M.; Kaushal, M.; Rabel, A.; Miller, J.L. Tough decoy targeting of predominant let-7 miRNA species in adult human hematopoietic cells. J. Transl. Med. 2017, 15, 169.
- Bluteau, O.; Langlois, T.; Rivera-Munoz, P.; Favale, F.; Rameau, P.; Meurice, G.; Dessen, P.; Solary, E.; Raslova, H.; Mercher, T.; et al. Developmental changes in human megakaryopoiesis. J. Thromb. Haemost. 2013, 11, 1730–1741.
- Stolla, M.C.; Catherman, S.C.; Kingsley, P.D.; Rowe, R.G.; Koniski, A.D.; Fegan, K.; Vit, L.; McGrath, K.E.; Daley, G.Q.; Palis, J. Lin28b regulates age-dependent differences in murine platelet function. Blood Adv. 2019, 3, 72–82.
- Battista, S.; Pentimalli, F.; Baldassarre, G.; Fedele, M.; Fidanza, V.; Croce, C.M.; Fusco, A. Loss of Hmga1 gene function affects embryonic stem cell lympho-hematopoietic differentiation. FASEB J. 2003, 17, 1496–8.
- Pierantoni, G.M.; Agosti, V.; Fedele, M.; Bond, H.; Caliendo, I.; Chiappetta, G.; Lo Coco, F.; Pane, F.; Turco, M.C.; Morrone, G.; et al. High-mobility group A1 proteins are overexpressed in human leukaemias. Biochem. J. 2003, 372, 145–150.
- Berent-Maoz, B.; Montecino-Rodriguez, E.; Fice, M.; Casero, D.; Seet, C.S.; Crooks, G.M.; Lowry, W.; Dorshkind, K. The expansion of thymopoiesis in neonatal mice is dependent on expression of High mobility group A 2 protein (Hmga2). PLoS One 2015, 10.
- Xi, Y.; Watanabe, S.; Hino, Y.; Sakamoto, C.; Nakatsu, Y.; Okada, S.; Nakao, M. Hmga1 is differentially expressed and mediates silencing of the CD4/CD8 loci in T cell lineages and leukemic cells. Cancer Sci. 2012, 103, 439–47.
- Nikolajczyk, B.S.; Nelsen, B.; Sen, R. Precise alignment of sites required for mu enhancer activation in B cells. Mol. Cell. Biol. 1996, 16, 4544–54.
- Nikolajczyk, B.S.; Cortes, M.; Feinman, R.; Sen, R. Combinatorial determinants of tissue-specific transcription in B cells and macrophages. Mol. Cell. Biol. 1997, 17, 3527–3535.
- Blackwell, T.K.; Moore, M.W.; Yancopoulos, G.D.; Suh, H.; Lutzker, S.; Selsing, E.; Alt, F.W. Recombination between immunoglobulin variable region gene segments is enhanced by transcription. Nature 1986, 324, 585–589.
- Sakai, E.; Bottaro, A.; Davidson, L.; Sleckman, B.P.; Alt, F.W. Recombination and transcription of the endogenous Ig heavy chain locus is effected by the Ig heavy chain intronic enhancer core region in the absence of the matrix attachment regions. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1526–31.
- Lewis, R.T.; Andreucci, A.; Nikolajczyk, B.S. PU.1-mediated transcription is enhanced by HMG-I(Y)-dependent structural mechanisms. J. Biol. Chem. 2001, 276, 9550–7.
- Nagulapalli, S.; Pongubala, J.M.; Atchison, M.L. Multiple proteins physically interact with PU.1. Transcriptional synergy with NF-IL6 beta (C/EBP delta, CRP3). J. Immunol. 1995, 155, 4330–8.
- McCarthy, K.M.; McDevit, D.; Andreucci, A.; Reeves, R.; Nikolajczyk, B.S. HMGA1 co-activates transcription in B cells through indirect association with DNA. J. Biol. Chem. 2003, 278, 42106–14.
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951.
- Dameshek W. Some speculations on the myeloproliferative syndromes [editorial]. Blood. 1951;6(4):372-375. Blood 2016, 127, 663.
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109.
- Rowley, J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293.
- Chen, C.C.; You, J.Y.; Lung, J.; Huang, C.E.; Chen, Y.Y.; Leu, Y.W.; Ho, H.Y.; Li, C.P.; Lu, C.H.; Lee, K. Der; et al. Aberrant let7a/HMGA2 signaling activity with unique clinical phenotype in JAK2-mutated myeloproliferative neoplasms. Haematologica 2017, 102, 509–518.
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148.
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790.
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061.
- Vainchenker, W.; Delhommeau, F.; Constantinescu, S.N.; Bernard, O.A. New mutations and pathogenesis of myeloproliferative neoplasms. Blood 2011, 118, 1723–1735.
- Aliano, S.; Cirmena, G.; Garuti, A.; Fugazza, G.; Bruzzone, R.; Rocco, I.; Malacarne, M.; Ballestrero, A.; Sessarego, M. HMGA2 overexpression in polycythemia vera with t(12;21)(q14;q22). Cancer Genet. Cytogenet. 2007, 177, 115–119.
- Storlazzi, C.T.; Albano, F.; Locunsolo, C.; Lonoce, A.; Funes, S.; Guastadisegni, M.C.; Cimarosto, L.; Impera, L.; D’Addabbo, P.; Panagopoulos, I.; et al. t(3;12)(q26;q14) in polycythemia vera is associated with upregulation of the HMGA2 gene [9]. Leukemia 2006, 20, 2190–2192.
- Etienne, A.; Carbuccia, N.; Adélaïde, J.; Bekhouche, I.; Rémy, V.; Sohn, C.; Sainty, D.; Gastaut, J.A.; Olschwang, S.; Birnbaum, D.; et al. Rearrangements involving 12q in myeloproliferative disorders: possible role of HMGA2 and SOCS2 genes. Cancer Genet. Cytogenet. 2007, 176, 80–88.
- Andrieux, J.; Demory, J.L.; Dupriez, B.; Quief, S.; Plantier, I.; Roumier, C.; Bauters, F.; Laï, J.L.; Kerckaert, J.P. Dysregulation and Overexpression of HMGA2 in Myelofibrosis with Myeloid Metaplasia. Genes Chromosom. Cancer 2004, 39, 82–87.
- Guglielmelli, P.; Zini, R.; Bogani, C.; Salati, S.; Pancrazzi, A.; Bianchi, E.; Mannelli, F.; Ferrari, S.; Le Bousse-Kerdilès, M.-C.; Bosi, A.; et al. Molecular Profiling of CD34 + Cells in Idiopathic Myelofibrosis Identifies a Set of Disease-Associated Genes and Reveals the Clinical Significance of Wilms’ Tumor Gene 1 ( WT1 ) . Stem Cells 2007, 25, 165–173.
- Young, A.R.J.; Narita, M. Oncogenic HMGA2: Short or small? Genes Dev. 2007, 21, 1005–1009.
- Ikeda, K.; Ogawa, K.; Takeishi, Y. The role of HMGA2 in the proliferation and expansion of a hematopoietic cell in myeloproliferative neoplasms. Fukushima J. Med. Sci. 2012, 58, 91–100.
- James, C.; Mazurier, F.; Dupont, S.; Chaligne, R.; Lamrissi-Garcia, I.; Tulliez, M.; Lippert, E.; Marion, F.X.; Pasquet, J.M.; Etienne, G.; et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood 2008, 112, 2429–2438.
- Ikeda, K.; Mason, P.J.; Bessler, M. 3’UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood 2011, 117, 5860–9.
- Li, O.; Li, J.; Dröge, P. DNA architectural factor and proto-oncogene HMGA2 regulates key developmental genes in pluripotent human embryonic stem cells. FEBS Lett. 2007, 581, 3533–3537.
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 131, 1109–1123.
- Ueda, K.; Ikeda, K.; Ikezoe, T.; Harada-Shirado, K.; Ogawa, K.; Hashimoto, Y.; Sano, T.; Ohkawara, H.; Kimura, S.; Shichishima-Nakamura, A.; et al. Hmga2 collaborates with JAK2V617F in the development of myeloproliferative neoplasms. Blood Adv. 2017, 1, 1001–1015.
- Oguro, H.; Yuan, J.; Tanaka, S.; Miyagi, S.; Mochizuki-Kashio, M.; Ichikawa, H.; Yamazaki, S.; Koseki, H.; Nakauchi, H.; Iwama, A. Lethal myelofibrosis induced by Bmi1-deficient hematopoietic cells unveils a tumor suppressor function of the polycomb group genes. J. Exp. Med. 2012, 209, 445–454.
- Yang, Y.; Akada, H.; Nath, D.; Hutchison, R.E.; Mohi, G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood 2016, 127, 3410–3423.
- Martin, S.E.; Sausen, M.; Joseph, A.; Kingham, B.F.; Martin, E.S. Identification of a HMGA2-EFCAB6 gene rearrangement following next-generation sequencing in a patient with a t(12;22)(q14.3;q13.2) and JAK2V617F-positive myeloproliferative neoplasm. Cancer Genet. 2012, 205, 295–303.
- Meyer, B.; Krisponeit, D.; Junghanss, C.; Escobar, H.M.; Bullerdiek, J. Quantitative expression analysis in peripheral blood of patients with chronic myeloid leukaemia: Correlation between HMGA2 expression and white blood cell count. Leuk. Lymphoma 2007, 48, 2008–2013.
- Wei, J.; Li, H.; Wang, S.; Li, T.; Fan, J.; Liang, X.; Li, J.; Han, Q.; Zhu, L.; Fan, L.; et al. let-7 enhances osteogenesis and bone formation while repressing adipogenesis of human stromal/mesenchymal stem cells by regulating HMGA2. Stem Cells Dev. 2014, 23, 1452–63.
- Bruchova, H.; Merkerova, M.; Prchal, J.T. Aberrant expression of microRNA in polycythemia vera. Haematologica 2008, 93, 1009–1016.
- Guglielmelli, P.; Tozzi, L.; Pancrazzi, A.; Bogani, C.; Antonioli, E.; Ponziani, V.; Poli, G.; Zini, R.; Ferrari, S.; Manfredini, R.; et al. MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp. Hematol. 2007, 35, 1708.e1-1708.e12.
- Zhan, H.; Cardozo, C.; Yu, W.; Wang, A.; Moliterno, A.R.; Dang, C. V.; Spivak, J.L. MicroRNA deregulation in polycythemia vera and essential thrombocythemia patients. Blood Cells, Mol. Dis. 2013, 50, 190–195.


