1000/1000
Hot
Most Recent
 +1 point
+1 point
Fabry disease (FD; OMIM#301500) is an X-linked lysosomal storage disorder associated with inherited or de novo disease causing variants in the α-galactosidase A gene (GLA; OMIM*300644). Reduced or even absent α-galactosidase A (α-Gal A; EC 3.2.1.22) activity leads to accumulation of glycosphingolipids with terminal α-D-galactosyl residues, especially globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) in plasma, urine and different organ systems, mainly cardiac, renal, endothelial and neuronal. The major physiological source of Gb3 is globoside, a glycolipid of erythrocytes and cells membranes found in different tissues.
Kidneys are very frequently affected in patients with Fabry disease regardless of gender. Most important manifestations of Fabry nephropathy are proteinuria and slowly progressive chronic kidney disease, which can in some cases lead to end stage renal disease.
FD is rare disorder with the estimated incidence in the general population between 1 in 40,000 and 1 in 117,000.[1] However, based on recent newborn screening studies, the prevalence in some populations was reported to be markedly higher with 1 in 1300 to 1 in 7800 males.[2]
Pathophysiological processes start at a very young age,[3] but most patients do not show symptoms in infancy. Gb3 deposition has been detected in fetuses[4] and in newborns with classical FD phenotype.[3] Men with classical phenotype, who have no or very low α-Gal A activity (<1% of mean normal), often develop first symptoms in early childhood, including acroparesthesias, angiokeratomas, intolerance to heat, hypo/anhidrosis, cornea verticillate and gastrointestinal disturbances.[5][6][7] The disease then gradually progresses to major organ failure in early adulthood, such as progressive kidney disease, cardiac signs, and symptoms and cerebrovascular events.[5][6][7] On the other hand, men who have a significant residual α-Gal A activity typically present with only one organ involvement.[6],[7] Although, FD is an X-linked trait, women can also develop typical symptoms due to random X-inactivation, which result in a mosaicism of gene expression, leading to differential expression of the functional or mutant enzyme.[8] Inactivation of the mutant allele leads to a milder phenotype, while the inactivation of the wild-type allele leads to more severe phenotype with an earlier onset.[9] The unaffected cells secrete mostly 46 kDa mature form of the α-Gal A and not the high-uptake mannose 6-phosphorilated form. The mature form is able to complement the activity in the population of cells lacking the expression of the enzyme.[10] Clinical presentation in women therefore varies from an asymptomatic or mild, later onset phenotype to a phenotype similar to that of men with a classical phenotype.[11]
The diagnosis of FD in men is based on reduced α-Gal A activity in plasma, leukocytes, or dried blood spots,[12][13][14] but in women, this measurement is unreliable, because enzyme activity in female with FD may not be elevated.[15] Gb3, the substrate of α-Gal A and the main accumulation deposit in FD, was considered as a possible diagnostic marker of FD. Gb3 accumulates mainly within cells, while it circulates in the extracellular space within lipoproteins.[16] However, there is no strict correlation between plasma Gb3 concentrations and clinical manifestation in Fabry patients.[17][18] Therefore, Gb3 level in urine and plasma are only suitable for identifying males with classical phenotype. Elevated plasma lyso-Gb3, the deacylated derivative of Gb3, has been designated as a hallmark of FD[19] and allows better differentiation between patients with classical and non-classical phenotype and healthy subjects.[20][21][22] Therefore, this biomarker may improve the diagnosis of clinically relevant FD, particularly in females with normal or borderline α-Gal A activity.[21] However, false negative results were also reported in non-classical phenotypes.[19][21] After the introduction of enzyme replacement therapy (ERT), patients with classical phenotypes show a rapid decrease in lyso-Gb3 concentration, whereas the decrease is slower in men with non-classical phenotype and women.[23][24] The serum lyso-Gb3 may serve as a marker for tissue involvement to assess which heterozygotes should be considered for treatment despite normal α-Gal A activity.[21] The gold standard for the diagnosis of female Fabry patients is still GLA gene sequencing, which is also important in men for the confirmation of the pathogenic GLA variant and prediction of the disease phenotype [28].[25] Early diagnosis of FD is important to initiate Fabry disease specific treatment, namely with ERT[24] or chaperones.[26]
Before the era of kidney transplantation and dialysis, end-stage renal disease (ESRD) was the leading cause of premature death in patients with FD.[1] However, progressive nephropathy remains one of the main manifestations of FD, which usually leads to ESRD in untreated patients with classical phenotype from the third to the fifth decade of life.[27][28] The rate of progression of chronic kidney disease (CKD) is similar to that of diabetic nephropathy.[29] The Fabry Outcome Survey reported a baseline prevalence of nephropathy in 59% of men and 38% of women with FD.[1] Even though progression to ESRD is less common in women with FD and are less likely to progress from moderate CKD to ESRD, the median age at which patients reached ESRD was 38 years regardless of their gender.[11][27] Renal involvement contributes largely to the overall burden of morbidity and mortality in FD.[6]
Gb3 accumulation occurs in various types of renal cells, including podocytes, mesangial, and interstitial cells, cells of the proximal and distal tubules, and loop of Henle, as well as vascular endothelial cells and smooth muscle cells.[30] The understanding of the pathophysiological mechanisms leading to CKD and ESRD is still scarce and further research is needed to clarify the complexity of genotype-phenotype correlation.
The main approaches used in nephrology to diagnose patients with FD are kidney biopsy, high-risk population testing, and family screening.[31] FD should be considered in patients with CKD without a clear cause of nephropathy.[2] For diagnosis, assessment of renal involvement, and monitoring of treatment, biomarkers of renal damage are often evaluated (albuminuria/proteinuria, serum creatinine, glomerular filtration rate (GFR), and cystatin C) together with urinary microscopy and renal biopsy.
Albuminuria/proteinuria is clinically most often used biomarker of Fabry nephropathy, although kidney damage is present already in the non-albuminuric state. Therefore they are not sensitive biomarkers for early kidney damage, as biopsies of normoalbuminuric Fabry patients have already shown advanced lesions.[32] However, proteinuria is one of the most important markers for monitoring the progression of nephropathy in treated and untreated patients, since patients with higher proteinuria show a much steeper decline in renal function over time.[27][29] Proteinuria has been shown to stimulate interstitial inflammation and fibrosis. Furthermore it promotes tubular cells to undergo partial epithelial mesenchymal trans-differentiation, which induces cell-cycle arrest and promotes the release of fibrogenic cytokines.[33][34]
Regular assessment of renal function in Fabry patients includes use of measured and estimated glomerular filtration rate (eGFR). Due to inaccuracy of creatinine-based GFR measurements, it is recommended to use measured GFR measurements (e.g., iohexol GFR) at least annually.[34] Because these methods are more complex and tedious, estimated GFR measurements using appropriate formula are more widely used. Currently used serum creatinine-based equations in the clinical practice are the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation for adults[35] and the Schwartz formula for children.[36] Some Fabry patients develop GFR loss before development of proteinuria.[11] Glomerular hyperfiltration may be a common feature in young Fabry patients and may be considered as an early marker of Fabry nephropathy,[37] which masks impairment of renal function, although rare, a decline in GFR can already be seen in adolescence.[38] Slope of progression of renal insufficiency was correlated to the level of proteinuria and in addition, it was not linear as shown by Schiffmann et al., where they retrospectively analyzed the decline of eGFR. The slope of renal function with an eGFR of more than 60 mL/min/1.73 m2 was −3.0 in men and −0.9 mL/min/1.73 m2/year in women, and with an eGFR of less than 60 mL/min/1.73 m2 it was −6.8 and −2.1 mL/min/1.73 m2/year in men and women, respectively.[39]
Cystatin-C, a cysteine protease inhibitor, is constantly produced by all nucleated cells. It is freely filtered by the glomerulus and then reabsorbed and catabolized in the tubular epithelial cells. Therefore, it does not re-enter the bloodstream or urine. Cystatin-C concentration was found as a superior and more sensitive marker than serum creatinine for detecting early renal dysfunction and small decreases in GFR in Fabry patients of both genders.[40] Therefore, it could be valuable as a prognostic marker and for estimating the efficiency of the ERT.[40] Probably the main reason why cystatin-C is not widely used is that it is more costly, time-consuming, and less available than creatinine.
As a non-invasive, reasonably priced, and expeditious diagnostic tool, urine microscopy can be useful for the diagnosis and assessment of FD progression. Most of the cells present in the urine of Fabry patients are renal tubular epithelial cells.[41] Mulberry cells with characteristic “Maltese cross bodies” (oval fat bodies) can be detected in the urinary sediments of Fabry patients under a polarized microscope.[42][43] Example of a Maltese cross in the urine sediment of FD patients is shown in Figure 1. Furthermore, there is a specific morphological population of Maltese cross bodies, characterized by a lamellarized appearance with protrusions, probably due to Gb3 resembling “mosquito coils”, which most likely represents a fragmentation of shed nephronal epithelial cells with accumulated lysosomal Gb3.[44] In addition their excretion correlated with the concentration of albumin in urine and could therefore be useful for accessing Fabry nephropathy burden.[44] Despite the fact, that this method could represent a valuable tool it is not commonly used, because it requires special equipment (phase contrast microscope) and well-trained personnel.

Figure 1. Birefringent Maltese cross in the urine sediment of Fabry patient when viewed under a polarized microscope (magnification 400×). Figure courtesy of Mravljak M; Department of Internal Medicine, General Hospital Slovenj Gradec.
Kidney biopsy with electron microscope analysis is recommended for all individuals with CKD, a GLA variant of unknown significance, and an uncertain diagnosis of FD to rule out other comorbidities, as it is currently the only reliable diagnostic method for confirming or excluding Fabry nephropathy.[45] Significant histologic changes occur before the typical clinical signs of CKD therefore, findings in biopsy are crucial for choosing the optimal therapeutic strategy and follow-up in high-risk patients.[46][47][48][49] Example of a kidney biopsy under electron microscopy with typical lamellar inclusions in a 27 year old Fabry male patient with normal kidney function (eGFR 102 mL/min/1.73 m2), normoalbuminuria (albumin to creatinine ratio (ACR) 12 mg/g), but high levels of podocyturia (urinary podocytes (uPod) 2420/g creatinine) is shown in Figure 2. Electron microscopic studies demonstrate typical osmophilic bodies, called myeloid or Zebra bodies, packed with lamellated membrane structures.[30] In contrast to electron microscopy, diagnosis could be much more challenging with light microscopy techniques, since the majority of stainings cause a washout of lipid contents. As a result, more unspecific vacuolization of podocytes and epithelial cells is a characteristic histological finding.[30][50] Mesangial expansion, segmental and global glomerulosclerosis, tubular atrophy, and intestinal fibrosis are also present at an early stage.[30][50][51] To better classify the extent of tissue involvement, the International Study Group of Fabry Nephropathy developed a scoring system of histological changes on light microscopy and toluidine blue–stained semithin sections.[49] The score quantifies the Gb3 density deposition in glomeruli, interstitium, and vessels, as well as progressive lesions (glomerulosclerosis, ischemic glomeruli, and tubulointerstitial fibrosis). Recently, bedside stereomicroscopy for Fabry kidney biopsies has been recommended as a complementary method to current histologic evaluation as it was demonstrated to have high diagnostic sensitivity for FD.[52]
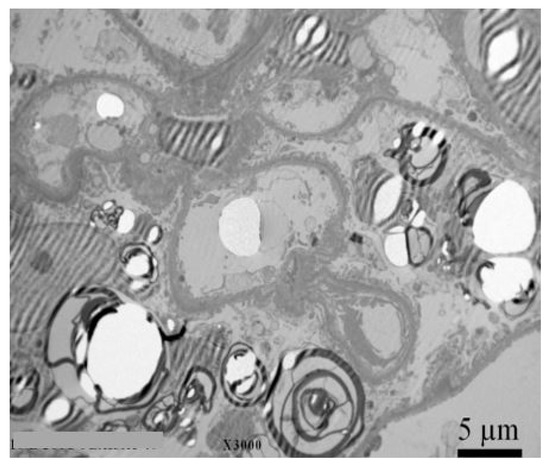
Figure 2. Kidney biopsy with electron microscopy with typical lamelar inclusions in a 27-year old Fabry male patient with normal kidney function (eGFR 102 mL/min/1.73 m2), normoalbuminuria (ACR 12 mg/g), but high levels of podocyturia (UPod 2.420/g creatinine). Diffuse and numerous myeloid inclusions in podocyte cytoplasm (black arrow) with presence of vacuoles in the cytoplasm of podocytes (white arrow) are evident. Figure courtesy of Pleško J and Kojc N; Institute of Pathology, Ljubljana Medical Faculty.
Heterogeneous phenotype, varying disease severity and symptoms onset make the diagnosis of FD notoriously difficult and often delayed for several years. Early diagnosis is crucial, as treatment should begin before the kidneys are irreversibly damaged. ERT was reported to reduce Gb3 deposits from most renal cells, but to a lesser extent in podocytes,[53] and to stop or slow down the progression of Fabry nephropathy.[54][55][56] However, once proteinuria occurs, it usually does not normalize despite treatment.[57] In addition, in patients with overt proteinuria or reduced GFR (<60 mL/min/1.73 m2), ERT does not prevent further deterioration of renal function.[2] Therefore, there is a considerable need for novel biomarkers that would enable identification and also prediction of development and progression of Fabry nephropathy.
Patients with rapid progression of kidney disease have higher urinary protein to creatinine ratio[58] and additionally, underlying conditions, such as hypertension, hyperlipidemia, and smoking, may contribute to a progressive loss of GFR.[59] However, environmental factors and glycolipid accumulation due to the disease causing GLA variants cannot fully explain the phenotypic variability. Even between the same family members carrying the same pathogenic GLA variant, there is enormous clinical variability.[60] It is therefore reasonable to anticipate that additional unknown biochemical, genetic, and epigenetic factors (modifiers) may influence the rate of progression of Fabry nephropathy. Recently, new biomarkers (bikunin, tubular proteins) have been proposed to improve the assessment of renal impairment, but further research is needed to evaluate their clinical utility.
Bikunin or urinary trypsin inhibitor is a serine protease inhibitor present in plasma and many tissues. It is excreted in urine and its levels were found significantly higher in Fabry patients with nephropathy; therefore, it may serve as a biomarker of renal impairment in FD.[61] The origin of the higher bikunin levels may imply direct renal involvement and secondary activation in response to the storage of glycosphingolipids in biochemical pathways associated with inflammation; however, further studies are needed to elucidate the mechanisms involved in the elevation of urinary bikunin levels.[61] Yet, renal impairment alone is not sufficient to explain higher urinary bikunin levels, as no correlation between serum creatinine and urinary bikunin levels was found.[61]
Impaired glomerular and tubular function in Fabry patients is reflected in an abnormal urinary excretion of tubular and glomerular proteins. Therefore, these biomarkers could be a valuable tool to assess kidney involvement and predict the progression of Fabry nephropathy. Larger studies are needed to thoroughly investigate the sensitivity and their correlations with Fabry nephropathy progression, the biomarkers currently used, and the changes in response to ERT.
Aguiar et al. found that the biomarkers of glomerular (transferrin and type IV collagen) and tubular (α1-microglobulin, N-acetyl-β-glucosaminidase, and alanine aminopeptidase) dysfunction were elevated even in a subgroup of patients without clinical signs of kidney disease. Furthermore, more significant correlation with eGFR was reported for type IV collagen and N-acetyl-β-glucosaminidase as it was for albuminuria.[62] Besides increased N-acetly-β-D-glucosaminidase, Shiffmann et al. also reported an increase in β2-microglobulin.[63] A decrease in the glomerular marker IgG, the tubular markers α1-microglobulin and retinol-binding protein as well as the shared tubular and glomerular markers albumin and transferrin in a population of 13 women with FD after long-term ERT was reported.[64] Furthermore another study showed normalization of urinary excretion of uromodulin after ERT and reduction in untreated Fabry patients.[65]
The earliest sign of renal damage appears to be podocyte foot process effacement, whereas it was found in young classic FD patients without clinically evident signs of Fabry nephropathy.[32][66][67] Podocytes as terminally differentiated cells do not divide; therefore, their replacement potential in adult is limited. Podocytes accumulate Gb3 more than other renal cell types.[30] Accumulation in podocytes continues until the third decade.[68] Alongside their volume continues to increase, resulting in an increase in podocyte foot process width and podocyte loss.[68] Injured podocytes detach from the glomerular basement membrane and are lost to urine. Podocyturia therefore correlates with the clinical severity of Fabry nephropathy.[67] A recent study of podocyte glycocalyx damage showed that podocalyxin loss may be associated with reduced adhesion of podocytes to the extracellular matrix, which enables detachment and urinary excretion.[69] Podocyturia disrupts glomerular permselectivity, causes albuminuria/proteinuria and leads to glomerulosclerosis and fibrosis.[70][71] A positive correlation was found between podocyturia and ACR.[72] Despite podocyturia is an early clinical sign of kidney injury and could serve as a diagnostic test to assess kidney involvement, it is still not regularly used in clinical practice. This is mainly because methods for assessing podocyturia are not yet standardized and not available in the majority of clinical settings.
Considering recent advances in sequencing approaches, it is reasonable to anticipate, that novel genomic and transcriptomics markers influencing the development and progression of the nephropathy are going to be identified. The metabolic origin and rate of progression of Fabry nephropathy resembles that of diabetic nephropathy. Since several studies have already been conducted in the field of CKD and diabetic nephropathy,[73] translating of the knowledge of biomarkers suspected of being involved in the development and progression of kidney failure could contribute to a better understanding of the molecular mechanisms of Fabry nephropathy. The development of new -omics technologies has led to new opportunities in the search for novel biomarkers. Despite several promising biomarkers, so far none of them have been translated into clinical practice. It is highly unlikely that the biomarkers discovered with -omics technologies will alone be sufficient to reliably predict the development and/or progression of Fabry nephropathy. Multifactorial models that would integrate clinical, genetic, and biochemical factors[74] are more likely to provide the evidence needed to translate this knowledge into clinical practice.


