 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Citarella | + 1147 word(s) | 1147 | 2020-09-08 06:28:06 | | | |
| 2 | Vicky Zhou | -75 word(s) | 1072 | 2020-09-27 07:41:19 | | | | |
| 3 | Vicky Zhou | Meta information modification | 1072 | 2020-10-27 08:52:35 | | |
Video Upload Options
Peptidyl fluoromethyl ketones occupy a pivotal role in the current scenario of synthetic chemistry, thanks to their numerous applications as inhibitors of hydrolytic enzymes. The insertion of one or more fluorine atoms adjacent to a C-terminal ketone moiety greatly modifies the physicochemical properties of the overall substrate, especially by increasing the reactivity of this functionalized carbonyl group toward nucleophiles.
1. Introduction
The main application of peptidyl α-fluorinated ketones in medicinal chemistry relies in their ability to strongly and selectively inhibit serine and cysteine proteases. These compounds can be used as probes to study the proteolytic activity of the aforementioned proteases and to elucidate their role in the insurgence and progress on several diseases. Likewise, if the fluorinated methyl ketone moiety is suitably connected to a peptidic backbone, it may confer to the resulting structure an excellent substrate peculiarity and the possibility of being recognized by a specific subclass of human or pathogenic proteases. Therefore, peptidyl fluoromethyl ketones are also currently highly exploited for the target-based design of compounds for the treatment of topical diseases such as various types of cancer and viral infections.
2. The Application of PFMKs in Drug Discovery
The development of peptidyl fluoromethyl ketones (PFMKs) has received over the last few decades a growing interest in drug discovery, since these types of compounds may be employed as substrates for a wide variety of biological targets [1]. The fluorinated electrophilic moieties of such substrates offer several advantages in comparison to other equivalent chemical units in terms of reactivity, selectivity, and therapeutic relevance. The mono-fluorinated function group, for instance, shows in general high reactivity and selectivity for cysteine proteases. At the same time, it exerts poor irreversible inhibition towards serine proteases and does not show any significant reactivity towards bionucleophiles such as glutathione [2]. On the other hand, a peptidic framework often represents the most foreseeable recognition motif for new biological targets and then the starting point of the target-based drug design. Usually, excellent substrate specificity can be fairly easily achieved by varying a single amino acid of the peptide sequence, with the position next to the electrophilic moiety (P1) being largely the most sensitive. As a matter of fact, various peptides bearing a C-terminal mono-fluoromethyl ketone (m-FMK) warhead have been used as selective activity-based probes for important druggable enzymatic targets, e.g., cathepsins (Cats), caspases (Casps), calpain I, SENPs, and N-glycanase. Unfortunately, their development as drugs has been compromised by the in vivo metabolic conversion of the m-FMK moiety into the highly toxic fluoroacetate [3]. In regard to the kinetics and mechanism of action of PFMKs, the high stability of the C–F bond was expected to lead a slow-binding reversible competitive inhibition of these nucleophilic targets (mostly hydrolytic enzymes) by electrophiles forming hemi(thio)ketal adducts (Figure 1). However, this has been ascertained for the majority of the tri-fluorinated derivatives (i.e., t-PFMKs) and very often observed in di-fluorinated derivatives (i.e., d-PFMKs), whereas it is well known that mono-fluorinated derivatives (i.e., m-PFMKs) are usually irreversible inhibitors leading to the formation of a covalent thioether adduct. The latter may be formed through the direct SN2 displacement of the fluoride group by the cysteine thiolate group or, more likely, through a two-step mechanism involving a thioemiketal and a three-membered sulfonium intermediate that instantly rearranges to afford the eventual thioether adduct (Figure 1) [4]. d-PFMKs and t-PFMKs, instead, have found a wide range of applications in synthetic medicinal chemistry, especially as enzyme inhibitors. The presence of additional fluorine atoms within their C-terminal warhead increases the electrophilicity of the FMK carbonyl group and makes them more susceptible to undergoing nucleophilic attack, also by the catalytic –OH group of serine proteases. Additionally, at variance with m-PFMKs, d-PFMKs and t-PFMKs do not present significant drawbacks related to the metabolism of the FMK moiety. These two fluorinated moieties exhibit another important characteristic; they can easily hydrate, depending on the nature of the amino acids they are bound to, forming very stable gem-diols which mimic the tetrahedral adduct of the transition-state of the enzyme-substrate hydrolysis mechanism (Figure 1). Therefore, the resulting PFMKs may also act exclusively as transition-state competitive analogues leading to rapidly reversible competitive inhibition [5][6]. In most cases, a mixture of the two FMK forms (i.e., carbonyl and hydrated) in different ratio is observed in solution, and a dual inhibition mechanism of the PFMKs is detected consequently.
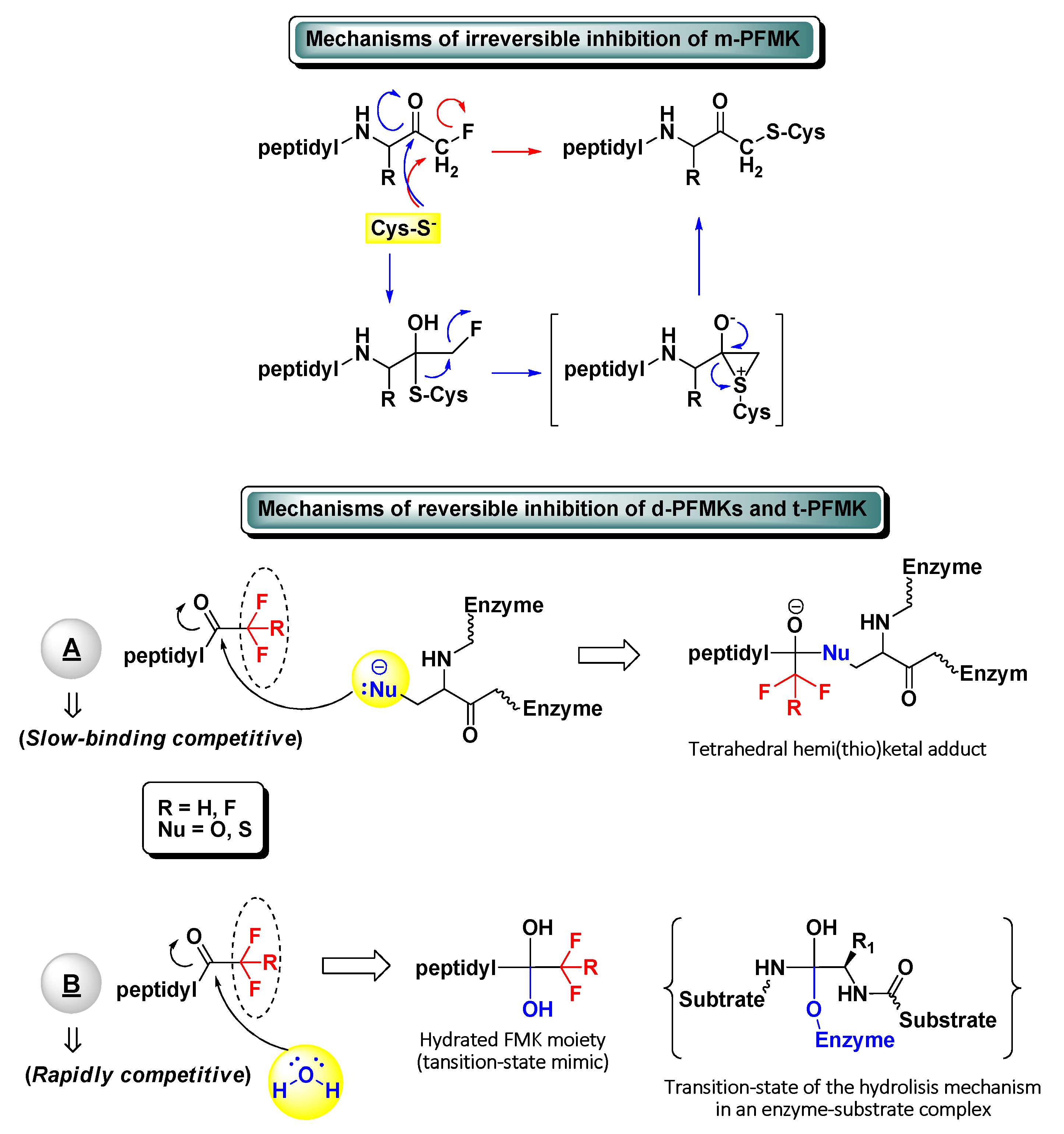
Figure 1. Schematic representation of the possible inhibitory mechanisms of action of all types of peptidyl fluoromethyl ketones (PFMKs).
3. Conclusion
Although the development of PFMKs originated more than fifty years ago, in particular with the mono-fluorinated derivatives employed as selective activity-based probes for the study of druggable enzymatic targets, the enormous potential of this type of compound remains rather unexplored. d-PFMKs and t-PFMKs are gaining considerable attention within the pharmaceutical research area, as both di-fluorinated and tri-fluorinated methyl ketone moieties are now recognized as valid electrophilic pharmacophores for the design of therapeutic agents. Compared to m-PFMKs, d-PFMKs and t-PFMKs are more prone to undergoing nucleophilic attack at the C-terminal warhead (thus they may target a wide variety of hydrolytic enzymes such as serine and cysteine proteases), are able to impart superior physicochemical properties to the mere peptidic substrate (e.g., lipophilia and binding selectivity), and do not present particularly relevant metabolic drawbacks. The fluorinated motifs of these two functional groups, i.e., -CHF2 and -CF3, may also serve as bioisosteres (-CH2OH and -CH2CH3, respectively) to impart specific steric and electronic properties to a compound or to prevent its metabolic degradation. The entire electrophilic synthons, instead, are widely exploited as critical intermediates in organic synthesis of bioactive molecules. Furthermore, the ability of the di-fluorinated and tri-fluorinated methyl ketone moieties to exist in a stable hydrated form (thus mimicking the hydrolysis transition-state and acting as reversible competitive analogues) expands the field of application of the related PFMKs to other enzymatic targets (e.g., aspartic proteases). Therefore, consistent efforts are also currently directed to the stereoselective synthesis of these electrophilic moieties joined to a peptidic substrate with the aim of obtaining PFMKs in the optically pure form at the P1 site, another important requirement to increase the target-specificity. The growing interest and popularity of the d-FMK and t-FMK moieties relies also on the recent outcomes and future perspectives related to the development of compounds bearing such functional groups and their ability to tackle efficaciously a variety of viral infections and tumors (which are currently the major cause of death worldwide) by inhibiting specific enzymatic targets.
References
- Barnes-Seeman, D.; Beck, J.; Springer, C. Fluorinated compounds in medicinal chemistry: Recent applications, synthetic advances and matched-pair analyses. Curr. Top. Med. Chem. 2014, 14, 855–864.
- Eichhold, T.H.; Hookfin, E.B.; Taiwo, Y.O.; De, B.; Wehmeyer, K.R. Isolation and quantification of fluoroacetate in rat tissues, following dosing of Z-Phe-Ala-CH2-F, a peptidyl fluoromethyl ketone protease inhibitor. J. Pharm. Biomed. Anal. 1997, 16, 459–467.
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.H.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38.
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750
- Rayo, J.; Munoz, L.; Rosell, G.; Hammock, B.D.; Guerrero, A.; Luque, F.J.; Pouplana, R. Reactivity versus steric effects in fluorinated ketones as esterase inhibitors: A quantum mechanical and molecular dynamics study. J. Mol. Model. 2010, 16, 1753–1764.
- Ngo, P.D.; Mansoorabadi, S.O.; Frey, P.A. Serine Protease Catalysis: A Computational Study of Tetrahedral Intermediates and Inhibitory Adducts. J. Phys. Chem. B 2016, 120, 7353–7359.


