 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura-Marinela AILIOAIE | + 3634 word(s) | 3634 | 2020-09-17 10:09:04 | | | |
| 2 | Nora Tang | -75 word(s) | 3559 | 2020-09-21 08:47:57 | | | | |
| 3 | Nora Tang | -75 word(s) | 3559 | 2020-09-21 08:52:00 | | |
Video Upload Options
Chronic arthritis is not a singular disorder; it is stands for multifaceted autoimmune and inflammatory disabling conditions causing joint pain or joint deformity. Chronic arthritis in children is diagnosed when all three of the following criteria are met. First, the age of onset is younger than 16 years. Second, arthritis includes swelling or effusion, or presence of two or more of the following: a) limitation of range of motion; b) tenderness or pain on motion; c) increased heat in one or more joints; and third, the duration of disease is 6 weeks or longer. Juvenile idiopathic arthritis (JIA) and adult rheumatoid arthritis (RA) are two major groups with chronic joint pain and inflammation, extra-articular manifestations, and high risk of comorbidities, which can cause physical and ocular disability, as well as create great socio-economic pressure worldwide.
The pathogenesis of arthritis manifested in childhood and adulthood is multifactorial, unclear, and overly complex, in which immunity plays an important role. Although there are more and more biological agents with different mechanisms of action for the treatment of arthritis, the results are not as expected, because there are partial responses or non-responsive patients to these compounds, high therapeutic costs, side effects, and so on; therefore, we must turn our attention to other therapeutic modalities. Updating knowledge on molecular and cellular mechanisms in the comparative pathogenesis of chronic arthritis in both children and adults is necessary in the early and correct approach to treatment. Photobiomodulation (PBM) represents a good option, offering cost-effective advantages over drug therapy, with a quicker, more positive response to treatment and no side effects. The successful management of PBM in arthritis is based on the clinician’s ability to evaluate correctly the inflammatory status of the patient, to seek the optimal solution, to choose the best technology with the best physical parameters, and to select the mode of action to target very precisely the immune system and the signaling pathways at the molecular level with the exact amount of quantum light energy in order to obtain the desired immune modulation and the remission of the disease. Light is a very powerful tool in medicine because it can simultaneously target many cascades of immune system activation in comparison with drugs, so PBM can perform very delicate tasks inside our cells to modulate cellular dysfunctions, helping to initiate self-organization phenomena and finally, healing the disease.
1. Introduction
Chronic arthritis is the most usual cause for joint pain, physical disability, and ocular invalidity worldwide. Juvenile idiopathic arthritis (JIA) and rheumatoid arthritis (RA) of the adult are two major groups with chronic joint inflammation, extra-articular manifestations, and high risk for comorbidities [1][2][3][4][5]. While in adults there are over 150 forms of chronic arthritis, in children, there are several dozen subtypes of the disease, but only juvenile polyarthritis with positive rheumatoid factor and the subtype of systemic arthritis also known as Still disease, which is more consistent with an autoinflammatory condition, have similar manifestations to adults [6].
Patients with systemic JIA require close keep under surveillance by a multidisciplinary team due to possible serious complications: macrophage activation syndrome, pericarditis, pulmonary hypertension, interstitial lung disease, infections, etc., which may be associated with increased mortality [7].
In the pathology of children, the oligoarticular manifestation is an entity that we do not find in the forms of rheumatic disease in adults and is characterized by often severe eye damage, localized growth disorders with elongation of a limb, and secondary posture disorders.
JIA is the type of chronic rheumatic disease that affects the child’s daily activities due to pain, joint swelling, morning stiffness, and locomotor and possibly ocular infirmities, which causes short-term and long-term disabilities, until adulthood and sometimes throughout life [8].
Treatment available for patients with chronic arthritis aims to reduce pain, maintain joint function, improve well-being, and prevent disability and associated comorbidities.
Pharmacological therapy usually includes non-steroidal anti-inflammatory drugs, intra-articular or systemic steroids, to which will be added disease-modifying anti-rheumatic drugs (DMARDs) and biological agents administered on time in the “window of opportunity” to prevent irreversible complications [2].
Early use of intra-articular steroid therapy, methotrexate, and biological agents introduced in recent decades have improved the prognosis of children with arthritis, but those with polyarticular form can have serious problems with active disease as adults. Most children with the JIA oligoarticular subtype may enter remission, but a small number progress to a persistent polyarticular form as adults.
Concerns have been raised about the use of biological agents that may increase the risk of cancer in patients with chronic arthritis.
Based on the severity of the disease, which evolves progressively, the patient with chronic arthritis can become an important burden for the family, but especially for the society, through the enormous costs of direct health care, social assistance, loss in education, productivity, and jobs.
The first goal of this review was to update knowledge on molecular and cellular mechanisms through a parallelism between special forms of chronic arthritis present in both children and adults, for an introspection into the pathogenesis of these diseases, in an attempt to reveal to researchers and clinicians the latest discoveries regarding new molecules and signaling pathways.
The second objective of this review was to raise awareness and send a signal to rheumatologists on the need to change the treatment paradigms for arthritis through innovative therapies to stop the perpetuation of the disease from childhood to adulthood, the side effects, the inefficiency in some cases, and the high current costs, in order to overcome this human and economic burden.
The third purpose was to promote light or laser therapies (photobiomodulation) as an important complementary and alternative method, which has become increasingly known around the world in recent decades for reducing pain and sometimes even eliminating the cause of the pain itself, for inducing early remission before common destructive changes in joints begin, in all arthritis forms.
Last aim but not least is to signal that photobiomodulation (PBM) and the single-cell live tracking technology of immune cell activities are ready to precisely target the signaling pathways and to find the answers to the complex interaction of the laser with the immune system, for “undoing” arthritis!
Seeking and developing new treatments to interact smoothly with the immune system both in children and adults to handle immune-mediated diseases that are becoming more and more complex is urgently needed.
JIA, formerly known as juvenile rheumatoid arthritis in the Anglo-Saxon literature, and chronic juvenile arthritis for French speakers, is a chronic immune-mediated inflammatory disease of unknown etiology and a complex genetic component that is defined according to the criteria of the International League of Associations for Rheumatology (ILAR) as inflammatory arthritis in one or more joints, which begins before the age of 16, persists for at least six weeks, and all other conditions that cause similar symptoms have been excluded [1][4].
A better understanding of the pathogenesis and the latest diagnostic tools are challenges for rheumatologists to update the classification. Based on ILAR criteria, there are seven main subgroups of JIA defined by clinical and laboratory data: systemic arthritis, rheumatoid factor (RF) polyarthritis—positive or negative, oligoarthritis (persistent or extensive), enthesitis-related arthritis (ERA), psoriatic arthritis (PsA), and a seventh category, undifferentiated arthritis, which includes those patients who do not fit any of the above forms of criteria [4][9].
2. Molecular and Cellular Mechanisms of Systemic Arthritis
The pathogenesis of systemic arthritis manifested in childhood and adulthood is multifactorial, unclear, and very complex, in which the innate immunity plays an important role by activating neutrophils and macrophages, as well as the adaptive immunity, by increasing the percentage of pro-inflammatory cytokines: interleukin (IL)-1β, IL-6, IL-18, and interferon gamma (IFN)–γ [10][11][12][13][14].
sJIA accounts for about 10% of all forms of juvenile arthritis and is a chronic disease that results in significant morbidity and mortality in children [15][16].
The most significant manifestation of systemic arthritis is its association with macrophage activation syndrome, a secondary disorder of excessive, uncontrolled activation and non-malignant proliferation of T lymphocytes and macrophages, with a state of hypercitokinemia, on which the clinical–biological signs depend [17][18][19].
The underlying cause of the occurrence of chronic rheumatism in the JIA subtypes, including sJIA, is largely unknown. A current concept would be that triggering manifestations in sJIA would be due to an infectious aggression with an inappropriate immune response due to a genetic or acquired immune defect [13]. More and more studies have shown that in the pathogenesis of sJIA, the innate immune system is more involved, compared to the adaptive one [11][20][21].
Biological studies for sJIA describe a polymorphism of disease-promoting elements encoded by tumor necrosis factor alpha (TNFα), IL-6, IL-10, macrophage migration inhibitory factor (MIF), and IL-1 family (in particular, IL1A, IL1RN, IL1R2) [22][23][24][25]. More and more clinical and biological as well as translational research draw attention to the particularly important role of IL-1β, IL-6, and IL-18 in the complexity of disease manifestation and the limited role of TNF-α, as well as the relative absence of induced chemokines by interferon gamma (IFN-γ), IFN-γ-inducible protein 10 (IP-10, CXCL10), MIG, and I-TAC [11][21][26]. IL-1 has a biphasic role in implicating innate immune mechanisms, but also in adaptive ones in triggering sJIA. Certain evidence of IL-1 involvement in the pathogenesis of sJIA is given by the successful treatment with IL-1 inhibitors, such as the biological agent anakinra, a soluble IL-1 receptor antagonist (IL-1Ra) that is similar to IL-1Ra, which has increased levels during the active disease observed also in polyarticular JIA. However, not all patients with sJIA respond well to anti-IL-1 therapy. The importance of IL-6 in the pathogenesis of sJIA has been demonstrated by correlating the serum and synovial concentration with the severe joint manifestations and the maximum level of fever [27]. Pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, by stimulating similar receptors, induce IL-6 production in lymphocytes, macrophages, and synovial cells [28].
IL-6 has various functions in the pathogenesis of sJIA in children and rheumatoid arthritis in adults, in the sense that it induces an acute phase response as well as activates immune reactions and hematopoiesis. The released IL-6 will induce the production of acute phase proteins (C-reactive protein and fibrinogen), which are known as biological markers of inflammation and the differentiation of naive T cells into Th17 cells [28][29].
The imbalance between Th17/Treg, where Th17 is activated significantly more than Treg, has a disastrous effect on RA development [30].
As IL-10 is a cytokine that would play a key anti-inflammatory role in the prevention of immune cascades from immune-mediated inflammatory diseases, conflicting results are reported in the literature regarding the poor involvement of this cytokine and the occurrence of manifestations in sJIA [24][25][31].
Both sJIA and macrophage activation syndrome are triggered by a cascade discharge of some cytokines such as interleukin 1β, IL-6 and IL-18.
To date, the exact role of interferon-gamma (IFN-γ), a cytokine with pro- and anti-inflammatory properties is being intensively investigated along with the role of NK cells providing IFN-γ [32].
In contrast, gene expression profiling was altered by the increased expression of innate genes, including TLR4 (Toll-Like Receptor 4) and S100A9 (S100 calcium-binding protein A9), and the decreased expression of immunity-regulating genes, such as IL-10RA (interleukin 10 receptor, alpha) and GZMK (granzyme K), as compared to cells from healthy controls. From these studies, it is believed that subtle defects in the pathways associated with NK cells, such as granzyme K expression and IFN-γ production determined by IL-18, may contribute to the immune aggregation of this disease [33].
3. Comparative Pathogenesis of Rheumatoid Arthritis in Adults and Children
The pathogenesis of RA and JIA is not yet very well known, although there is strong evidence that it involves the components of the immune system, especially T and B lymphocytes, as well as the antibodies and cytokines resulting from this immune conflict [7][34].
In rheumatoid arthritis, the activation of a naive T cell departing from the thymus to the lymphoid organs involves coordinated interactions between a number of molecules on the surface of this cell and an antigen-presenting cell (APC), that is, that carries an antigenic peptide derived from the infectious agent noncovalently linked to a major histocompatibility complex (MHC) class I or class II molecule.
When the APC cell is activated, various costimulatory ligands are expressed, allowing the activation, proliferation, and differentiation of T cells [35][36].
The T cells will express a series of inhibitory receptors for a fine regulation of the response appropriate to the inflammatory environment where they were being stimulated. Inhibitory receptors can act in two directions: to limit the costimulatory signaling, as well as to temporarily bind the costimulatory molecule [35].
At the synovial level, as result of inflammation, the differentiation of naive T cells in Th17 cells will occur. It was believed that this immune pathology would be mediated by Th1 cells (the first objectified), but today, the research has evolved, and it has been discovered that, in fact, Th17 cells are considered responsible for the pathogenesis in rheumatoid arthritis [37][38].
Current studies demonstrate that synovial fibroblasts and activated immune cells are directly involved in the production and release of many pro-inflammatory cytokines that play a crucial role in the development and progression of RA [39].
In fact, the characteristic inflammatory process in RA is achieved by the abundance of inflammatory-promoting cytokines, in counterbalance with inhibitory cytokines, intercellular communication, immune responses, and boosting cell movement to territories of inflammatory, infectious, or post-traumatic conflict.
In RA, the cytokines of the immune network are classified into four groups: pro-inflammatory cytokines, inflammatory cytokines in the joints, anti-inflammatory cytokines, and natural cytokine antagonists [40].
After the onset of initial stimuli, the cytokines play an important role in communicating with the components of the immune system at each stage of the pathophysiology of RA. The release of cytokines, particularly the TNF-α, IL-6, and IL-1, promotes the synovial inflammatory process.
Recent clinical studies have shown that patients with RA (adults) or active polyarticular sJIA (children), who did not respond adequately to MTX (methotrexate) and TNF-alpha inhibitors, received an IL-6 inhibitor, for example, tocilizumab in children and sarilumab (in adults), which are biologics that can be more effective [41][42].
Among the pro-inflammatory cytokines at the synovial level, TNF-α is a pleiotropic cytokine produced by several cell types, such as T and B cells, but also by innate immune cells (dendritic, monocyte, neutrophil, mast cells) and has a very important role, because it participates as the main mediator in regulating and training other factors [43][44]. In addition to this role, it is known that TNF-α is associated with bone and cartilage destruction by activating chondrocytes and osteoclasts [45]. TNF-α induces the synthesis and secretion of MMPs (matrix metalloproteinases), which in turn affect the chemokine and cytokine action of MMP-2, MMP-3, MMP-7, and MMP-9, which release TGF beta (Transforming Growth Factor) from the matrix, thus enabling its activation [46]. Therefore, anti-TNF biological therapy has been considered a remarkable breakthrough in the treatment of chronic autoimmune diseases, such as RA and JIA [43].
The interleukin (IL)-1 (family) together with its members (IL-33, IL-36α, β, γ, IL-37, and IL-38), IL-6, and IL-12 superfamilies (IL-27, IL-35) together with the other key cytokines (IL-15, IL-16, IL-17 family IL-17A, IL-17B, IL-17C), the recently cloned cytokine IL-18, IL-32, IL-34, and interferon (IFN)-y, the granulocyte macrophage colony-stimulating factor, are detected in a high concentration in the synovial fluid, but also in the patient’s serum, thus leading to the process of local joint destruction and systemic effects in the rheumatoid arthritis patient [40][47].
More explicitly, IL-1 has 11 pro-inflammatory and anti-inflammatory members, which are chronologically numbered based on their discovery, from the IL-1 first family member 1 (IL-1F1) to IL-1F11. More commonly, they are also known as receptor antagonist IL-1α, IL-1β, IL-1 (IL-1Ra), IL-18, IL-33, IL-36α, IL-36β, IL-36γ, IL-36Ra, IL-37, and IL-38 [48].
IL-33 has been detected in high serum concentrations in adult patients with rheumatoid arthritis, in contrast to those with osteoarthritis (OA) and psoriatic arthritis (PsA) and was associated with bone erosion and cardiovascular pathology, as a predictive factor for the evolution of atherosclerotic plaque [47]; however, the results are contradictory for its real role in the pathogenesis of RA, and as a consequence, specific drugs are not yet available [49][50][51][52].
IL-17A has a direct influence on the early pathogenesis and chronic stages of synovitis in rheumatoid and psoriatic arthritis, through systemic, but also local effects on keratinocytes [53].
It has been shown that in the synovial lymphocyte infiltration and in the hyperplastic mucosa of RA, there are cells producing IL-17A and IL-17F; at the same time, there is a recruitment of Th17 cells that will interact with local cells and perpetuate chronic inflammation [54].
IL-17 is directly involved in the stimulation of vascular endothelial growth factor production in synovial fibroblasts, angiogenesis, and synovial pannus development [55][56]. The interaction between Th17 cells and synoviocytes is crucial, because of this cooperation, IL-17 will be massively released [53]. TNF-alpha supports the effect of human IL-17A for the action of increasing the secretion of IL-6 and IL-8 from rheumatoid synoviocytes and vice versa, IL-17A and IL-17F induce TNFα receptor II expression and production [53][57][58].
Another particularly important role of IL-17 is to promote the expression of nuclear factor kappa-B (NF-κB) ligand receptor activator (RANKL) on osteoblasts and synoviocytes and to activate RANK signaling in osteoclasts [59][60][61].
The pro-inflammatory cytokines are also responsible for the synthesis of chemokines from MMPs, inducible nitric-oxide synthase, osteoclasts differentiation, and an increased expression of cell adhesion molecules. Disruption of MMP activity can lead to tissue degradation associated with inflammation in rheumatoid arthritis.
Helper T cells are deeply involved in the pathogenesis of autoimmune diseases, including RA; for example, it has been shown recently that Th17 can move into a “non-classical” class of Th1.
Today, it is known that Th17 produces the cytokine IL-17 [28], which activates inflammation by stimulating immune cells and at the same time activates osteoclasts by inducing kappa B ligand nuclear factor activator receptor (RANKL) in synovial fibroblasts. This fact opens new horizons for Th17-targeted therapies to stop the bone destruction associated with T cell activation [62].
At the same time, Foxp3 is essential for the suppressive function of Treg cells, and as a specific marker of Th17 cells, it accelerates osteoclasts differentiation. In RA, Foxp3(+)CD4(+) T cells are subjected to conversion into Th17 cells, which is mediated by synovial fibroblast-derived IL-6 [110], and meanwhile, IFN-gamma cytokines, IL-4, and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), produced by Th1, Th2, and respectively, Treg, regulate osteoclast differentiation. In RA, there is an imbalance in the Th17/Treg ratio, where Th17 is activated much more than Treg [63]. It is speculated that TNF inhibitors used in RA therapy reduce the passage of Th17 cells to non-classical Th1 cells, as well as direct inhibit the TNFα [43][64]. Inflammatory synovitis both in RA and JIA provides the image of an imbalance between pro-inflammatory and anti-inflammatory cytokines [IL-10, IL-11, and IL-13], which are insufficient to counterbalance the intensely active inflammatory process.
Although there are more and more biological agents with different mechanisms of action for the treatment of rheumatoid arthritis in children and adults, the results are not as we expected, because there are partial responses or non-responsive patients to these compounds, high therapeutic costs, side effects, and so on; therefore, we must turn our attention to other therapeutic modalities to induce disease remission.
4. New Introspections and Perspectives on Photobiomodulation in Arthritis
As an interdisciplinary field, photomedicine is growing in importance because of its relevance to light and laser therapies [65]. Applied photobiomodulation could be a safe and an exceptionally good option in the multidisciplinary management of rheumatoid arthritis and chronic pain in children and adults.
To achieve the desired effect of photobiomodulation, a certain quantified amount of photonic energy is always required to target the cells and the immune signaling pathways, to modulate the immune system, and LASERS, LEDs or other available light devices can be used accordingly.
There is currently no consensus on the effective PBM treatment method in improving symptoms and remission of chronic rheumatic diseases.
Successful management of PBM in arthritis is based on the clinician’s ability to evaluate correctly the inflammatory status of the patient, to seek the optimal solution, to choose the best technology with the best physical parameters and mode of action, so that at molecular level the treatment can target very precisely the immune system and the molecular signaling pathways with the exact amount of quantum light energy in order to obtain the desired immune modulation and the remission of the disease.
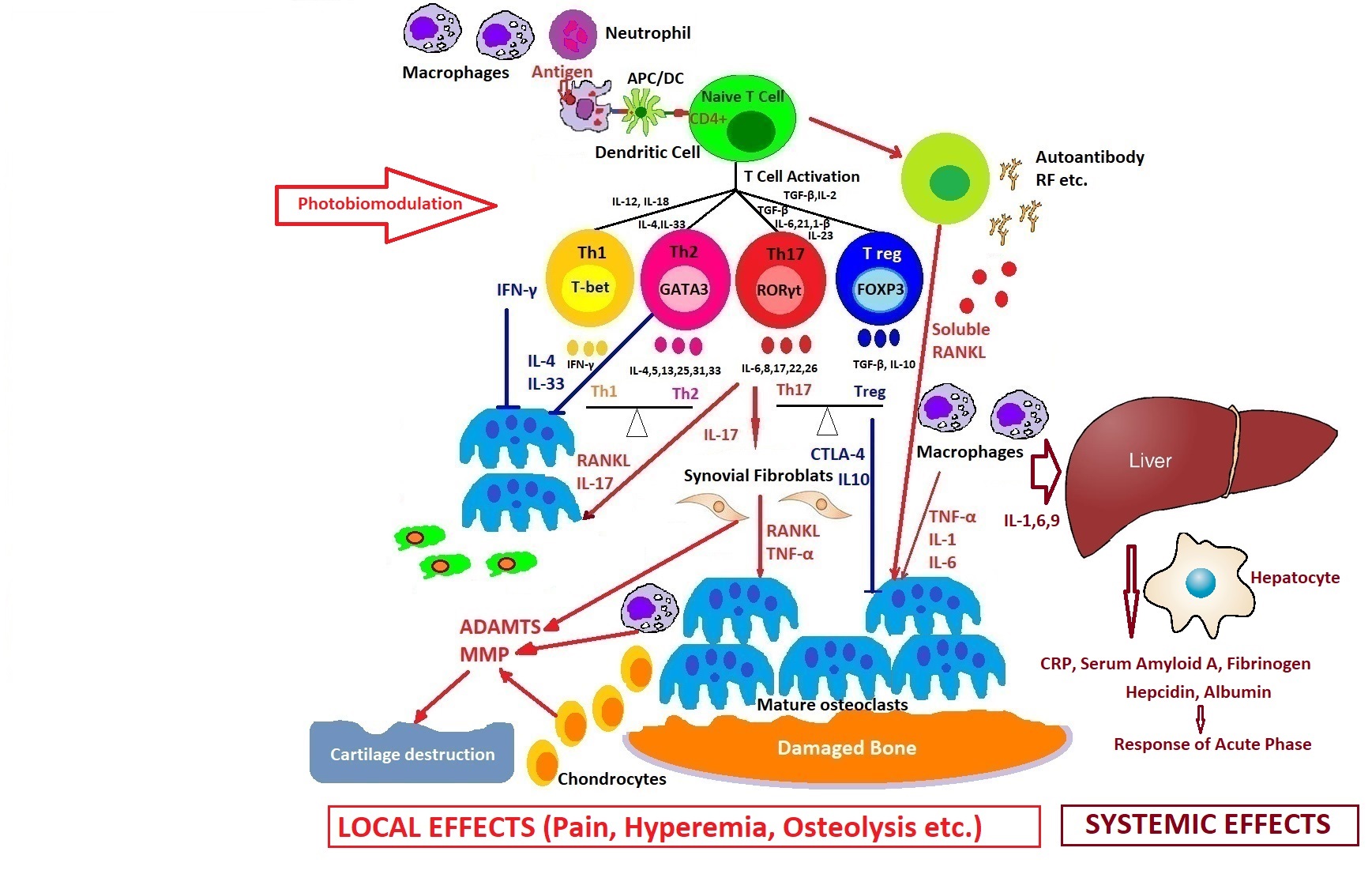
How photobiomodulation could regulate the immune response in arthritis. Possible mechanisms of action on excessive T cell immune response, regulation of pro- and anti-inflammatory cytokines balance, and the process of stopping the proliferative synovium and the osteocartilaginous destruction.
Light is a very powerful tool in medicine because it can simultaneously target many cascades of immune system activation in comparison with drugs, so PBM can perform very delicate tasks inside our cells to modulate cellular dysfunctions, helping to initiate self-organization phenomena and finally healing the disease.
A lot of information can be stored or transmitted using light. The near future will be focused on state-of-the-art laser therapy, in an atmosphere concentrated not only on reducing pain and inflammation, but also early healing of the disease.
Interdisciplinary teams should work diligently to meet these needs by also using single-cell imaging devices for multispectral laser photobiomodulation on immune cells.
A new field of innovative research with multiple treatment options in immune-mediated inflammatory diseases opens up by the application of photobiomodulation with important clinical implications for the future.
References
- Sherry, D.D.; Bhaskar, R.S.A.; Poduval, M.; Rabinovich, C.L.; Myones, B.L. Juvenile Idiopathic Arthritis. Available online: https://emedicine.medscape.com/article/1007276-overview (accessed on 24 August 2020).
- Ailioaie, C.; Ailioaie, L.M. Juvenile idiopathic arthritis. In Management of Chronic Rheumatic Pain; PIM Publishing House: Iaşi, Romania, 2008; pp. 129–146.
- Haasnoot, A.J.W.; Sint Jago, N.F.M.; Tekstra, J.; de Boer, J.H. Impact of Uveitis on Quality of Life in Adult Patients with Juvenile Idiopathic Arthritis. Arthritis Care Res. 2017, 69, 1895–1902.
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis: Second Revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392.
- Cassidy, J.T.; Kivlin, J.; Lindsley, C.; Nocton, J. Ophthalmologic Examinations in Children with Juvenile Rheumatoid Arthritis. Pediatrics 2006, 117, 1843–1845.
- World Health Organization. Chronic rheumatic conditions. Available online: https://www.who.int/chp/topics/rheumatic/en/ (accessed on 19 August 2020).
- Systemic juvenile idiopathic arthritis: Clinical manifestations and diagnosis. Available online: https://www.uptodate.com/contents/systemic-juvenile-idiopathic-arthritis-clinical-manifestations-and-diagnosis (accessed on 19 August 2020).
- Angelis, A.; Kanavos, P.; López-Bastida, J.; Linertová, R.; Serrano-Aguilar, P.; BURQOL-RD Research Network. Socioeconomic costs and health-related quality of life in juvenile idiopathic arthritis: A cost-of-illness study in the United Kingdom. BMC Musculoskelet. Disord. 2016, 17, 321.
- Aggarwal, R.; Ringold, S.; Khanna, D.; Neogi, T.; Johnson, S.R.; Miller, A.; Brunner, H.I.; Ogawa, R.; Felson, D.; Ogdie, A.; et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res. 2015, 67, 891–897.
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486.
- Woo, P. Systemic juvenile idiopathic arthritis: Diagnosis, management, and outcome. Nat. Clin. Pract. Rheumatol. 2006, 2, 28–34.
- Mellins, E.D.; Macaubas, C.; Grom, A.A. Pathogenesis of systemic juvenile idiopathic arthritis: Some answers, more questions. Nat. Rev. Rheumatol. 2011, 7, 416–426.
- Prakken, B.; Albani, S.; Martini, A. Juvenile idiopathic arthritis. Lancet 2011, 377, 2138–2149.
- Nigrovic, P.A.; Schneider, R. Systemic Juvenile Idiopathic Arthritis and Adult Onset Still Disease. In Textbook of Autoinflammation; Hashkes, P., Laxer, R., Simon, A., Eds.; Springer: Cham, Switzerland, 2019; pp. 587–616.
- Cassidy, J.T.; Petty, R.E. Chronic Arthritis in Childhood. In Textbook of Pediatric Rheumatology, 5th ed.; Cassidy, J.T., Petty, R.E., Laxer, R., Lindlsy, C., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2005; pp. 206–321.
- Singh-Grewal, D.; Schneider, R.; Bayer, N.; Feldman, B.M. Predictors of disease course and remission in systemic juvenile idiopathic arthritis: Significance of early clinical and laboratory features. Arthritis Rheum. 2006, 54, 1595–1601.
- Kelly, A.; Ramanan, A.V. Recognition and management of macrophage activation syndrome in juvenile arthritis. Curr. Opin. Rheumatol. 2007, 19, 477–481.
- Weaver, L.K.; Behrens, E.M. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr. Opin. Rheumatol. 2014, 26, 562–569.
- Put, K.; Avau, A.; Brisse, E.; Mitera, T.; Put, S.; Proost, P.; Bader-Meunier, B.; Westhovens, R.; Van den Eynde, B.J.; Orabona, C.; et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: Tipping the balance between interleukin-18 and interferon-γ. Rheumatol. (Oxf. Engl.) 2015, 54, 1507–1517.
- Ramanan, A.V.; Grom, A.A. Does systemic-onset juvenile idiopathic arthritis belong under juvenile idiopathic arthritis? Rheumatol. (Oxf.) 2005, 44, 1350–1353.
- Barnes, M.G.; Grom, A.A.; Thompson, S.D.; Griffin, T.A.; Pavlidis, P.; Itert, L.; Fall, N.; Sowders, D.P.; Hinze, C.H.; Aronow, B.J.; et al. Subtype-specific peripheral blood gene expression profiles in recent-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009, 60, 2101–2112.
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J. Clin. Invest. 1998, 102, 1369–1376.
- Ogilvie, E.M.; Fife, M.S.; Thompson, S.D.; Twine, N.; Tsoras, M.; Moroldo, M.; Fisher, S.A.; Lewis, C.M.; Prieur, A.-M.; Glass, D.N.; et al. The -174G allele of the interleukin-6 gene confers susceptibility to systemic arthritis in children: A multicenter study using simplex and multiplex juvenile idiopathic arthritis families. Arthritis Rheum. 2003, 48, 3202–3206.
- Fife, M.S.; Gutierrez, A.; Ogilvie, E.M.; Stock, C.J.; Samuel, J.M.; Thomson, W.; Mack, L.F.; Lewis, G.M.; Woo, P. Novel IL10 gene family associations with systemic juvenile idiopathic arthritis. Arthritis Res. Ther. 2006, 8, R148.
- Möller, J.C.; Paul, D.; Ganser, G.; Range, U.; Gahr, M.; Kelsch, R.; Rösen-Wolff, A.; Hedrich, C.M. IL10 promoter polymorphisms are associated with systemic onset juvenile idiopathic arthritis (SoJIA). Clin. Exp. Rheumatol. 2010, 28, 912–918.
- Fall, N.; Barnes, M.G.; Thornton, S.; Luyrink, L.; Olson, J.; Ilowite, N.T.; Gottlieb, B.S.; Griffin, T.; Sherry, D.D.; Thompson, S.; et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007, 56, 3793–3804.
- De Benedetti, F.; Massa, M.; Robbioni, P.; Ravelli, A.; Burgio, G.R.; Martini, A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991, 34, 1158–1163.
- Tateiwa, D.; Yoshikawa, H.; Kaito, T. Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review. Cells 2019, 8, 818.
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636.
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517.
- Muller, K.; Herner, E.B.; Stagg, A.; Bendtzen, K.; Woo, P. Inflammatory cytokines and cytokine antagonists in whole blood cultures of patients with systemic juvenile chronic arthritis. Br. J. Rheumatol. 1998, 37, 562–569.
- Vandenhaute, J.; Avau, A.; Filtjens, J.; Malengier-Devlies, B.; Imbrechts, M.; Van den Berghe, N.; Ahmadzadeh, K.; Mitera, T.; Boon, L.; Leclercq, G.; et al. Regulatory Role for NK Cells in a Mouse Model of Systemic Juvenile Idiopathic Arthritis. J. Immunol. 2019, 203, 3339–3348.
- Put, K.; Vandenhaute, J.; Avau, A.; Van Nieuwenhuijze, A.; Brisse, E.; Dierckx, T.; Rutgeerts, O.; Garcia-Perez, J.; Toelen, J.; Waer, M.; et al. Inflammatory gene expression profile and defective IFN-gamma and granzyme K in natural killer cells of systemic juvenile idiopathic arthritis patients. Arthritis Rheumatol. 2017, 69, 213–224.
- Fournier, C. Where do T cells stand in rheumatoid arthritis? Joint Bone Spine 2005, 72, 527–532.
- Pennock, D.N.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283.
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995.
- Kasama, T.; Isozaki, T.; Takahashi, R.; Miwa, Y. Clinical effects of tocilizumab on cytokines and immunological factors in patients with rheumatoid arthritis. Int. Immunopharmacol. 2016, 35, 301–306.
- Burmester, G.R.; Feist, E.; Dörner, T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 77–88.
- Ponchel, F.; Goëb, V.; Parmar, R.; El-Sherbiny, Y.; Boissinot, M.; El Jawhari, J.; Burska, A.; Vital, E.M.; Harrison, S.; Conaghan, P.G.; et al. An immunological biomarker to predict MTX response in early RA. Ann. Rheum. Dis. 2014, 73, 2047–2053.
- Nalbant, S.; Birlik, A.M. Cytokines in Rheumatoid Arthritis (RA). In New Developments in the Pathogenesis of Rheumatoid Arthritis; Sakkas, L.I., Ed.; IntechOpen: Rijeka, Croatia, 2017; Available online: https://www.intechopen.com/books/new-developments-in-the-pathogenesis-of-rheumatoid-arthritis/cytokines-in-rheumatoid-arthritis-ra- (accessed on 25 August 2020).
- Yokota, S.; Tanaka, T.; Kishimoto, T. Efficacy, safety and tolerability of tocilizumab in patients with systemic juvenile idiopathic arthritis. Ther. Adv. Musculoskelet. Dis. 2012, 4, 387–397.
- Gabay, C.; Msihid, J.; Zilberstein, M.; Paccard, C.; Lin, Y.; Graham, N.M.H.; Boyapati, A. Identification of sarilumab pharmacodynamic and predictive markers in patients with inadequate response to TNF inhibition: A biomarker substudy of the phase 3 TARGET study. RMD Open 2018, 4, e000607.
- Davignon, J.; Rauwel, B.; Degboé, Y.; Constantin, A.; Boyer, J.F.; Kruglov, A.; Cantagrel, A. Modulation of T-cell responses by anti-tumor necrosis factor treatments in rheumatoid arthritis: A review. Arthritis Res. Ther. 2018, 20, 229.
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology 2012, 51, 3–11.
- Jimenez-Boj, E.; Redlich, K.; Türk, B.; Hanslik-Schnabel, B.; Wanivenhaus, A.; Chott, A.; Ramiro, S.; Schett, G. Interaction between synovial inflammatory tissue and bone marrow in rheumatoid arthritis. J. Immunol. 2005, 175, 2579–2588.
- Alamgeer, H.U.; Uttra, A.M.; Qasim, S.; Ikram, J.; Saleem, M.; Niazi, Z.R. Phytochemicals targeting matrix metalloproteinases regulating tissue degradation in inflammation and rheumatoid arthritis. Phytomedicine 2020, 66, 153134.
- Alunno, A.; Carubbi, F.; Giacomelli, R.; Gerli, R. Cytokines in the pathogenesis of rheumatoid arthritis: New players and therapeutic targets. BMC Rheumatol. 2017, 1, 3.
- Dinarello, C.; Arend, W.; Sims, J.; Smith, D.; Blumberg, H.; O’Neill, L.; Goldbach-Mansky, R.; Pizarro, T.; Hoffman, H.; Bufler, P.; et al. IL-1 family nomenclature. Nat. Immunol. 2010, 11, 973.
- Matsuyama, Y.; Okazaki, H.; Tamemoto, H.; Kimura, H.; Kamata, Y.; Nagatani, K.; Yoshio, T.; Nagashima, T.; Iwamoto, M.; Hayakawa, M.; et al. Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J. Rheumatol. 2010, 37, 18–25.
- Hong, Y.S.; Moon, S.J.; Joo, Y.B.; Jeon, C.H.; Cho, M.L.; Ju, J.H.; Oh, H.J.; Heo, Y.J.; Park, S.H.; Kim, H.Y.; et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J. Korean Med. Sci. 2011, 26, 1132–1139.
- Xiangyang, Z.; Lutian, Y.; Lin, Z.; Liping, X.; Hui, S.; Jing, L. Increased levels of interleukin-33 associated with bone erosion and interstitial lung diseases in patients with rheumatoid arthritis. Cytokine 2012, 58, 6–9.
- Talabot-Ayer, D.; McKee, T.; Gindre, P.; Bas, S.; Baeten, D.L.; Gabay, C.; Palmer, G. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine 2012, 79, 32–37.
- Robert, M.; Miossec, P. IL-17 in Rheumatoid Arthritis and Precision Medicine: From Synovitis Expression to Circulating Bioactive Levels. Front. Med. 2019, 5, 364.
- Eljaafari, A.; Tartelin, M.L.; Aissaoui, H.; Chevrel, G.; Osta, B.; Lavocat, F.; Miossec, P. Bone marrow-derived and synovium-derived mesenchymal cells promote Th17 cell expansion and activation through caspase 1 activation: Contribution to the chronicity of rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2147–2157.
- Honorati, M.C.; Neri, S.; Cattini, L.; Facchini, A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage 2006, 14, 345–352.
- Daoussis, D.; Andonopoulos, A.P.; Liossis, S.N. Wnt pathway and IL-17: Novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin. Arthritis Rheum. 2010, 39, 369–383.
- Hot, A.; Miossec, P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann. Rheum. Dis. 2011, 70, 727–732.
- Hot, A.; Zrioual, S.; Toh, M.L.; Lenief, V.; Miossec, P. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann. Rheum. Dis. 2011, 70, 341–348.
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713.
- Van Bezooijen, R.L.; Papapoulos, S.E.; Löwik, C.W. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: Is there dependency on nuclear factor-kappaB and receptor activator of nuclear factor kappaB (RANK)/RANK ligand signaling? Bone 2001, 28, 378–386.
- Lavocat, F.; Maggi, L.; Annunziato, F.; Miossec, P. T-cell clones from Th1, Th17 or Th1/17 lineages and their signature cytokines have different capacity to activate endothelial cells or synoviocytes. Cytokine 2016, 88, 241–250.
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682.
- Takayanagi, H. Osteoimmunology and the effects of the immune system on bone. Nat. Rev. Rheumatol. 2009, 5, 667–676.
- Kotake, S.; Yago, T.; Kobashigawa, T.; Nanke, Y. The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. J. Clin Med. 2017, 6, 67.
- Ailioaie, L.M.; Ailioaie, C. Photobiomodulation—Targeting the quantum life. Newest implications for immunity, health, and youth. Invited Lecture. In Proceedings of the 11th International ISLA Congress for Medical Laser Applications, Lauenförde-Beverungen, Germany, 10–11 June 2016.


