 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salihanur Darici | + 4458 word(s) | 4458 | 2020-09-17 10:46:29 | | | |
| 2 | Camila Xu | Meta information modification | 4458 | 2020-09-21 03:10:49 | | | | |
| 3 | Camila Xu | Meta information modification | 4458 | 2020-09-23 08:54:58 | | | | |
| 4 | Camila Xu | Meta information modification | 4458 | 2020-09-23 09:02:22 | | | | |
| 5 | Camila Xu | Meta information modification | 4458 | 2020-09-23 09:56:20 | | |
Video Upload Options
Acute myeloid leukemia (AML) is a highly heterogeneous hematopoietic malignancy, characterized by excessive proliferation and accumulation of immature myeloid blasts in the bone marrow. While reductions of bulk malignant cells can be achieved in the majority of patients by standard chemotherapy consisting of cell cycle active drugs, such as cytarabine and anthracyclines, approximately two-thirds of patients relapse after the induction therapy, highlighting an unmet need for a more targeted therapeutic approach. A rare population of therapy-resistant cells are believed to be the origin of relapse, termed leukemia stem cells (LSCs), also referred to as leukemia-initiating cells (LICs). These cells acquire enhanced self-renewal capacity and exhibit a block in differentiation.
1. Definition
Acute myeloid leukemia (AML) is a highly heterogeneous hematopoietic malignancy, characterized by excessive proliferation and accumulation of immature myeloid blasts in the bone marrow [1]. While reductions of bulk malignant cells can be achieved in the majority of patients by standard chemotherapy consisting of cell cycle active drugs, such as cytarabine and anthracyclines, approximately two-thirds of patients relapse after the induction therapy, highlighting an unmet need for a more targeted therapeutic approach [2]. A rare population of therapy-resistant cells are believed to be the origin of relapse, termed leukemia stem cells (LSCs), also referred to as leukemia-initiating cells (LICs) [3][4][5]. These cells acquire enhanced self-renewal capacity and exhibit a block in differentiation.
2. Introduction
The phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathway emerges as a promising therapeutic candidate to sensitize LSCs to chemotherapy. It plays an important role in both normal and malignant hematopoiesis; components of this pathway govern the expression of genes and proteins essential for cell proliferation, differentiation, and survival. Constitutive activation of PI3K/Akt/mTOR pathway is detected in 50–80% of AML patients, associated with decreased overall survival (OS) [6][7][8]. Mutations in receptor tyrosine kinases (RTKs) or GTPases are the major causes leading to upregulation of the PI3K/Akt/mTOR pathway in AML [9]. One important mechanism leading to deregulation of PI3K/Akt/mTOR signaling is mutation of fms-like tyrosine kinase 3 (FLT3). Among them, internal tandem duplication (ITD) of FLT3 gene (FLT3-ITD) is one of the most frequent mutations in normal karyotype AML (approximately 25%). In recent clinical studies, few patients display prolonged remissions with RTK inhibitors, such as FLT3 inhibitors, highlighting the need for novel and/or partner targeted therapies [10][11]. Targeting the PI3K/Akt/mTOR pathway may be an option for FLT3-ITD AML patients.
Hyperactivation of PI3K/Akt/mTOR has also been associated with attenuated sensitivity to chemotherapy. Several studies have demonstrated that PI3K/Akt/mTOR inhibition may preferentially target LSCs. For example, the PI3K/Akt/mTOR pathway may regulate LSC survival through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The pro-inflammatory transcription factor NF-κB, has been found to be aberrantly activated in LSCs but is not expressed in normal human CD34+ progenitor cells [12][13][14]. NF-κB is known to mediate chemoresistance by upregulation of anti-apoptotic genes, which enable cells to increase proliferation and evade apoptosis [15][16][17]. Targeting NF-κB may be selective for LSCs and/or sensitize LSCs to chemotherapy. Notably, NF-κB is a downstream target of PI3K/Akt/mTOR, and this signaling cascade can trigger NF-κB activation, which suggests a common survival pathway for LSCs. Treatment of AML patient samples with PI3K inhibitor LY294002 displayed inhibited Akt phosphorylation and NF-κB DNA-binding activity [18]. Furthermore, PI3K/Akt/mTOR inhibition induced apoptosis in primary AML cells and potentiated response to cytotoxic chemotherapy, while sparing normal HSC function, [19][20][21][22]. The LSC population was targeted by PI3K-directed therapies demonstrated by reduced engraftment ability of these cells in nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice [19][23]. PI3K/Akt/mTOR inhibitors may additionally potentiate LSC kill by synergizing with LSC-directed therapies. An essential feature of quiescent AML-LSCs is that they have relative lower production of reactive oxygen species (ROS) compared with bulk cells [24]. These ROS-low LSCs were shown to aberrantly overexpress Bcl-2, making them more susceptible to eradication by small-molecule Bcl-2 inhibitors like venetoclax. The therapeutic potential of venetoclax could be enhanced by PI3K/Akt/mTOR inhibition through Mcl-1-dependent mechanisms, which is a well-known determinant of resistance to venetoclax [25].
Growing evidence signposts PI3K as a druggable target for AML; indeed, there has been very productive development of small-molecule inhibitors targeting the PI3K/Akt/mTOR pathway. While PI3K/Akt/mTOR inhibitors have been effective treating other hematological malignancies, such as chronic lymphoblastic leukemia (CLL) and follicular lymphoma (FL), in AML, the clinical potential of PI3K/Akt/mTOR inhibitors has not yet been fully elucidated [26][27][28]. Clinical studies using PI3K/Akt/mTOR inhibitors as monotherapy have shown limited therapeutic efficacy, likely due to compensatory activation of other survival pathways [29].
3. The PI3K/Akt/mTOR Signaling Pathway
3.1. Regulation of the PI3K/Akt/mTOR Pathway in Normal Hematopoiesis
The PI3K family consists of three distinct classes of PI3Ks (I-III), of which class I is implicated in regulation of hematopoiesis. Class I PI3K can be further divided into class IA and class IB enzymes, both of which are activated by cell surface receptors. Class IA PI3K can be activated by RTKs, G protein-coupled receptors (GPCRs), and oncoproteins such as the small G protein Ras, whereas class IB PI3K can be activated by GPCRs only [30][31]. Class IA PI3Ks form heterodimers between one of three catalytic subunits (p110α, p110β, or p110δ) and a regulatory adaptor molecule (p85α (or its splice variants p55α and p50α), p85β or p55γ) [32][33]. Each pair shares some overlap whilst maintaining distinct function. In contrast to the heterogeneity of class IA, a single class IB isoform has been described that associates catalytic subunit p110γ with regulatory adaptor molecule p101 or p84 [34][35]. While catalytic subunits p110α and p110β are consistently expressed in a broad range of tissues, p110γ and p110δ are specifically enriched within the hematopoietic system—preferentially in leukocytes [36].
In response to extracellular stimuli (e.g., hormones, growth factors, and cytokines) and the subsequent activation of RTKs, class IA PI3K is recruited to the plasma membrane via interaction of p85 with adaptor proteins, such as insulin receptor substrate (IRS) 1/2 or growth factor receptor-bound protein 2-associated binding protein 2 (GAB2) that bind to the regulatory p85 subunit of PI3K [37][38]. The class IB p110γ is activated by GPCRs through direct interaction of its regulatory adaptor molecule with Gβγ subunit of trimeric G proteins [35]. The activated p110 catalytic subunit facilitates the phosphorylation of phosphatidylinositol-4,5-phosphate (PIP2) to generate phosphatidylinositol-3,4,5-phosphate (PIP3). PIP3 recruits phosphoinositide-dependent kinase 1 (PDK1) and Akt/protein kinase B (PKB) to the plasma membrane where PDK1 phosphorylates Akt at Threonine(T)308 residue within the activation loop of the kinase domain to initiate the activation of Akt [39][40] (Figure 1).
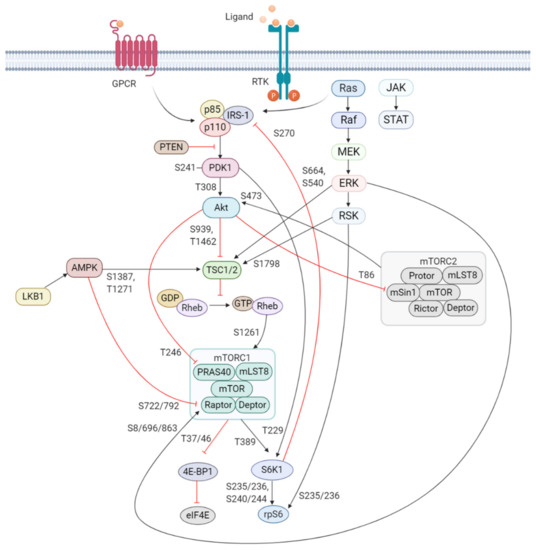
Figure 1. Schematic overview of the activation and regulation of the PI3K/Akt/mTOR signaling pathway. Activation of PI3K is stimulated by binding of an extracellular ligand (e.g., hormones, growth factors, and cytokines) to a cell surface receptor such as the receptor tyrosine kinase (RTK) in the plasma membrane. Activated RTK recruits adaptor proteins, which bind to the regulatory p85 subunit of PI3K and subsequently activate the catalytic subunits for full PI3K activation. PI3K is also activated by G protein-coupled receptors (GPCR) or small GTPase Ras, which bind PI3K directly. Activated PI3K catalyzes the phosphorylation of phosphatidylinositol-4,5-phosphate (PIP2) to generate phosphatidylinositol-3,4,5-phosphate (PIP3). PIP3 recruits phosphoinositide-dependent kinase 1 (PDK1) and Akt to the plasma membrane inducing Akt phosphorylation by PDK1 at T308. Akt activation is completed by phosphorylation at S473 by mTOR complex 2 (mTORC2). The mTOR complex includes two distinct protein complexes, mTORC1 and mTORC2. mTORC1 comprises of mTOR, proline-rich Akt substrate 40 kDa (PRAS40), regulatory-associated protein of mTOR (Raptor), mammalian lethal with Sec13 protein 8 (mLST8, also known as GβL), and DEP-domain-containing mTOR-interacting protein (Deptor) [41]. mTORC2 comprises of mTOR, mLST8, Deptor, protein observed with Rictor-1 (Protor), rapamycin-insensitive companion of mTOR (Rictor), and mammalian stress-activated protein kinase interacting protein (mSin1) [42]. Akt indirectly activates mTORC1 by phosphorylation and inhibition of tuberous sclerosis complex 2 (TSC2) at S939 and T1462, releasing the inhibitory effects of this complex on Ras-related GTPase Rheb, an activator of mTORC1. Akt also directly controls activation of mTORC1 in a TSC2-independent manner via phosphorylation of PRAS40 at T246. The extracellular signal-regulated kinase (ERK)/90 kDa ribosomal S6 kinase (RSK) and liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) signaling pathways impinge on several nodes of the PI3K/Akt/mTOR pathway and can modulate mTORC1 activity. Both ERK and RSK modulate mTORC1 activity by phosphorylation of TSC2 at S664 and S540 (ERK) and S1798 (RSK). ERK1/2 can also control mTORC1 activation by phosphorylation of Raptor at S8, S696, and S863. Master metabolic regulator AMPK inhibits mTORC1 activity in two different pathways, the first by phosphorylation of TSC2 at T1271 and S1387 and the second by phosphorylation of Raptor at S722 and S792. Activated mTORC1 promotes cap-dependent mRNA translation via phosphorylation of eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) at T37 and T46, which is a priming event required for subsequent phosphorylation of several carboxy-terminal serum-sensitive sites to release 4E-BP1 from eIF4E. Ribosomal protein S6 kinase beta-1 (S6K1) is a downstream target of mTORC1, activated by phosphorylation at T389 by mTORC1 as well as T229 phosphorylation mediated by PDK1. S6K1 in turn activates ribosomal protein S6 (rpS6), which is dispensable for cell growth and protein synthesis. RSK can also directly activate rpS6 via phosphorylation at S235 and S236. The black arrows represent positive regulation (activation), whereas the red blunt-ended lines indicate negative regulation (inhibition). IRS-1 = insulin receptor substrate 1, PTEN = phosphatase and tensin homolog, GDP = guanosine diphosphate, GTP = guanosine triphosphate, JAK = Janus kinase, STAT = signal transducer and activator of transcription. Created with BioRender.com.
Akt is a highly conserved serine/threonine kinase that has multiple diverse functions. Full Akt activation, in addition to PDK1-mediated phosphorylation, requires phosphorylation at Serine(S)473 residue in the regulatory domain, by mTOR complex 2 (mTORC2), integrin-linked kinase (ILK), PDK1 or members of the PI3K-related kinase (PIKK) family, such as DNA-dependent protein kinase (DNA-PK) [43][44]. Notably, Akt can activate mTORC2 through a positive feedback loop by direct phosphorylation of mTORC2 component mammalian stress-activated protein kinase interacting protein (mSin1) at T86 [45][46]. Activated Akt can phosphorylate a wide spectrum of protein substrates, including forkhead box class O (FoxO), glycogen synthase kinase-3 (GSK3) α/β, and Bcl-2 associated agonist of cell death (BAD), maintaining cell cycling, survival, metabolism, cell growth and other essential cellular functions (Figure 2) [47].
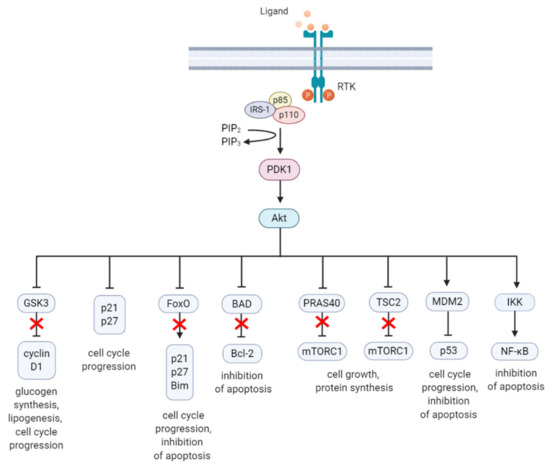
Figure 2. A summary diagram of Akt downstream target molecules. (From left to right) Fully activated Akt controls numerous effectors implicated in cell growth, proliferation, differentiation, metabolism, and survival, of which some are highlighted. Akt regulates G1/S cell cycle progression by phosphorylation and inactivation of GSK3/cyclin D1, p21, and p27. Akt was found to regulate cell metabolism by mediating lipogenesis and glucose uptake through phosphorylation and inhibition of GSK3, which inhibits glycogen synthesis. Akt controls apoptosis by phosphorylation and inhibition of FoxO and pro-apoptotic Bcl-2 family member BAD. Akt promotes cell growth by activation of mTORC1 though phosphorylation of PRAS40, which prevents its inhibition of mTORC1. Akt can also induce mTORC1 activation through phosphorylation and inhibition of TSC2, relieving the inhibitory effects of the TSC1/TSC2 complex on mTORC1. Akt enhances MDM2-mediated ubiquitination and proteasomal-dependent degradation of p53. Akt can inhibit apoptosis and promote cell survival by activating NF-κB. The arrows represent positive regulation (induction/activation), whereas the blunt-ended lines indicate negative regulation (inhibition/inactivation). The red cross represents inhibition caused by Akt-mediated negative regulation. RTK = receptor tyrosine kinase, IRS-1 = insulin receptor substrate 1, PIP2 = phosphatidylinositol-4,5-phosphate, PIP3 = phosphatidylinositol-3,4,5-phosphate, PDK1 = phosphoinositide-dependent kinase-1, MDM2 = mouse double minute 2 homolog, GSK3 = glycogen synthase kinase 3, TSC2 = tuberous sclerosis complex 2, mTORC1 = mTOR complex 1, FoxO = forkhead box O, Bim = Bcl-2-like protein 11, BAD = BCL-2 associated agonist of cell death, Bcl-2 = B-cell lymphoma 2, IKK = IκB kinase, NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells. Created with BioRender.com.
Regulation of PI3K activity is negatively controlled by the tumor suppressor phosphatase and tensin homolog (PTEN) and Src homology domain-containing inositol phosphatase (SHIP) to maintain normal hematopoiesis. PTEN is a lipid phosphatase that hampers PI3K signaling through dephosphorylation of the lipid signaling intermediate PIP3 [48]. Loss of function of PTEN through mutations, genetic silencing, or epigenetic mechanism is implicated in the pathology of multiple human malignancies and can lead to aberrant PI3K/Akt/mTOR signaling [49]. SHIP is predominantly expressed in hematopoietic cells and hydrolyzes PIP3 to generate PI(3,4)P2 [50]. Mutation of the SHIP gene was detected in a low percentage of AML and acute lymphoblastic leukemia (ALL) patients [51].
One of the key downstream targets of the Akt is mTOR, which positively regulates cell growth and proliferation by promoting protein synthesis and inhibition of autophagy [52][53]. The identification of this serine/threonine kinase stems from the discovery of the natural product rapamycin, originally extracted from the soil bacterium Streptomyces hygroscopicus [54]. mTOR participates in two functionally distinct multiprotein complexes, mTORC1 and mTORC2, of which only mTORC1 is sensitive to inhibition by rapamycin [55]. Akt indirectly activates mTORC1 by phosphorylation of tuberous sclerosis complex 2 (TSC2) at S939 and T1462 [56][57]. TSC2 functions with TSC1 forming a heterodimeric complex which blocks the activation of Ras homolog enriched in brain (Rheb). Akt phosphorylation of TSC2 inhibits the GTPase-activating protein (GAP) activity of this complex and in turn permits Rheb to activate mTORC1. Akt can also activate mTORC1 by a TSC2-independent mechanism which involves phosphorylation of proline-rich Akt substrate 40kDa (PRAS40), a component of mTORC1. Phosphorylation of PRAS40 at T246 results in dissociation of PRAS40 from mTORC1 and attenuates the inhibitory effect of PRAS40 on mTORC1 activity [58]. Upon activation, mTORC1 is phosphorylated on several residues (T2446, S2448, and S2481), but no function has been ascribed to any phosphorylation site [59][60][61]. S1261 was identified as a site-specific phosphorylation site of mTORC1 that in response to insulin signals via the PI3K/TSC2/Rheb axis regulating mTORC1 function in an amino acid-dependent and rapamycin-insensitive mechanism.
The highly conserved protein kinase mTOR is a central hub of nutrient signaling and cell growth and integrates multiple intracellular signals [62]. With respect to the mTOR signaling pathway, the TSC1/TSC2 complex has emerged as a sensor and integrator of multiple signaling pathways to modulate mTORC1 activity [63] (Figure 1). One important signaling pathway that negatively regulates mTORC1 activity is the liver kinase B1 (LKB1)/ AMP-activated protein kinase (AMPK) pathway [64]. AMPK is a cellular energy sensor activated in various conditions that deplete cellular energy, such as nutrient deprivation or hypoxia [65][66]. Phosphorylation of AMPK at T172 in the activation loop is required for its kinase activity and is mediated by LKB1, the upstream serine/threonine kinase of AMPK [67]. AMPK inhibits mTORC1 in two different ways, i.e., phosphorylation of TSC2 at T1227 and S1345; and phosphorylation of Raptor at S722/792, which is a component of mTORC1 [68][69]. In addition to AMPK, the extracellular signal-regulated kinase (ERK)/ p90 ribosomal S6 kinase (RSK) pathway also modulates mTORC1 activity. ERK/RSK is one of the other main signaling networks activated in parallel with PI3K by RTKs to control survival, differentiation, proliferation, and metabolism [70]. Both ERK and RSK promote mTORC1 activity by phosphorylation of TSC2 at ERK S664 and S540, and RSK S1798 [71][72]. In addition, ERK1/2 contributes to Ras-dependent activation of mTORC1 through phosphorylation of Raptor at S8, S696, and S863 [73].
Upon activation, mTORC1 phosphorylates its main downstream targets eukaryotic initiation factor-4E (eIF4E) -binding protein 1 (4E-BP1) and rapamycin-sensitive ribosomal protein S6 kinase beta-1/p70 ribosomal S6 kinase (S6K1), involved in the translation of mRNAs. 4E-BP1 inhibits the initiation of cap-dependent translation by binding and inactivating eIF4E. This binding is reversible, and mTORC1 phosphorylation of 4E-BP1 at T37/46 relieves 4E-BP1 from eIF4E [74]. Released eIF4E assembles at the 5′ end of mRNA, which facilitates the recruitment of the ribosome and subsequent initiation of translation [75].
S6K1 is the other main target of mTORC1 implicated in the regulation of cell growth. Activation of S6K1 requires phosphorylation at T229 and T389, of which T229 is phosphorylated by PDK1 and T389 by mTORC1 [76][77][78]. Activated S6K1 activates 40S ribosomal protein (rp) S6 that represents the most extensively studied substrate of S6K1. rpS6 becomes phosphorylated on several serine residues [79]. While S6K1 phosphorylates rpS6 on all phosphorylation sites (S235/236 and S240/244), RSK phosphorylates rpS6 exclusively on S235/236 in an mTOR-independent mechanism, suggesting that ERK/RSK pathway contributes to rpS6 phosphorylation upon mitogen stimulation [80]. mTORC1-stimulated S6K1 mediates an important negative feedback regulation of PI3K through phosphorylation of IRS-1. As such, S6K1 phosphorylates IRS-1 proteins at several serine residues (S270, S307, S636, and S1101) of which S270 was found to be required for S6K1/IRS-1 interaction and subsequent phosphorylation of the other S6K1-specific residues [81]. Phosphorylation of IRS-1 induces its protein degradation and insulin resistance, thereby inhibiting the insulin-like growth factor 1 (IGF-1) -mediated PI3K activation.
3.2. Constitutive PI3K/Akt/mTOR Activation in AML
Regulated PI3K/Akt/mTOR signaling is critical for normal hematopoiesis, with deregulation of PI3K/Akt/mTOR activity linked to depletion of HSC pool [82]. For example, Pten deletion in adult mice HSCs activated the PI3K/Akt/mTOR pathway and promoted HSC proliferation and depletion through induced expression of p16Ink4a and p53, and leukemogenesis [83][84][85]. These effects were mostly mediated by mTOR as rapamycin was able to suppress leukemogenesis and restore normal HSC function [85]. Myristoylated Akt1 (myr-Akt) was introduced into HSCs via retroviral transduction of bone marrow cells and subsequent transplantation, to mimic constitutively active Akt, which is frequently observed in AML [82]. Results revealed that myr-Akt contributes to myeloproliferative disorders (MPD), and T-cell lymphoma with high frequency, and AML with a lower penetrance. HSCs in the myr-Akt1 mice displayed transient expansion of immature myeloid cells in the bone marrow and spleen, and increased cycling, associated with impaired engraftment. The importance of mTOR signaling as a mediator of Akt was demonstrated with rapamycin. Rapamycin rescued cobblestone formation in myr-Akt–transduced bone marrow cells in vitro and increased survival of myr-Akt mice. TSC1 is also potentially involved in leukemogenesis. Deletion of TSC1 in HSCs leads to activation of mTOR signaling, causes rapid HSC cycling and elevated levels of ROS, and impaired HSC self-renewal [86]. Importantly, treatment with a ROS antagonist in vivo demonstrated that the TSC1/mTOR axis is important to maintain HSC quiescence and function by suppressing ROS. These findings indicate that mTOR is an important mediator of PI3K regulation in HSCs. Furthermore, FoxO transcription factors are functionally redundant in HSC homeostasis through regulation of HSC response to physiologic oxidative stress, quiescence, and survival [87][88][89]. Mice engineered with conditional knockout alleles of Foxo1, Foxo3, and/or Foxo4 displayed increased cell cycling and apoptosis of HSC, and a marked increase in ROS levels.
The PI3K/Akt/mTOR signaling pathway is frequently hyperactivated in AML cells and potentially contributes to uncontrolled growth, proliferation, differentiation, metabolism, and survival [90][91]. The PI3K/Akt/mTOR pathway is also important for the regulation of the AML-LSC population, demonstrated in mouse models with genetic alterations of key PI3K/Akt/mTOR signaling genes. Rheb1 is overexpressed in AML patients, which was associated with reduced survival in comparison to patients with lower Rheb1 expression [92][93][94]. Deletion of Rheb1 induced apoptosis and enhanced cell cycle arrest in LSCs, and prolonged survival of MLL-AF9-induced leukemic mice, suggesting that the mTORC1 pathway may be required for LSC maintenance [95]. PDK1 is overexpressed in over 40% of AML patients and is required for Akt activation [96]. Deletion of Pdk1 in MLL-AF9-induced mice resulted in a reduction of LSCs and upregulated the expression of apoptosis inducers, such as BAX and Tp53 [97][98]. Mice transplanted with MLL-AF9-positive LSCs were also shown dependent on S6K1 for LSC maintenance [99]. Loss of S6K1 improved survival of MLL-AF9-induced leukemic mice, which was associated with reduced Akt and 4E-BP1 phosphorylation. Furthermore, the PI3K/Akt/mTOR pathway is also implemented in the crosstalk between LSCs and the stromal cells associated with its niche that promotes the drug-resistant phenotype of LSCs. Several reports have demonstrated that pharmacological inhibition of PI3K/Akt/mTOR signaling may effectively target leukemic cells within the bone marrow niche [100][101][102].
Dysregulation of the PI3K/Akt/mTOR pathway is often the result of loss or inactivation of tumor suppressors, mutation or amplification of PI3K, as well as activation of RTKs or oncoproteins upstream of PI3K (Figure 3) [103][104]. About 50–80% of patients with AML display constitutive PI3K/Akt/mTOR activation, and this was associated with significant poorer OS [105]. Poor prognosis in AML patients with constitutive PI3K/Akt/mTOR signaling could be related to the fact that this pathway is associated with chemoresistance, which contributes to the short-term survival in AML [106][107][108]. However, no correlation was shown to exist between PI3K/Akt/mTOR activity and a particular AML subtype, cytogenetic abnormality, or etiology of the disease [109][110].
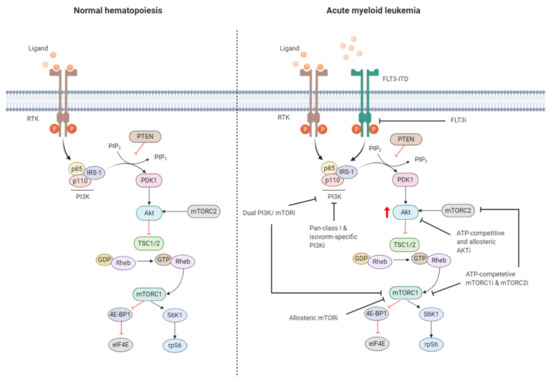
Figure 3. Targeting the PI3K/Akt/mTOR pathway in AML. The PI3K/Akt/mTOR pathway is commonly dysregulated in AML caused by mutations in membrane-bound proteins such as receptor tyrosine kinases (RTKs) and small GTPase Ras. Activating mutations in fms-like tyrosine kinase 3 (FLT3), such as the FLT3-internal tandem duplication (FLT3-ITD), are an important mechanism leading to dysregulation of PI3K/Akt/mTOR signaling. The ITD mutation causes ligand-independent activation of the FLT3 receptor, leading to constitutive activation of the PI3K/Akt/mTOR pathway. Numerous small-molecule inhibitors of this pathway include FLT3 inhibitors (FLT3i), dual PI3K/mTORi, allosteric mTORi, pan-class I and isoform-specific PI3Ki, ATP-competitive and allosteric Akti, and ATP-competitive mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) inhibitors (mTORC1i and mTORC2i). The red arrow indicates elevated Akt phosphorylation, whereas the red blunt-ended lines represent negative regulation (inhibition). IRS-1 = insulin receptor substrate 1, PIP2 = phosphatidylinositol-4,5-phosphate, PIP3 = phosphatidylinositol-3,4,5-phosphate, PTEN = phosphatase and tensin homolog, PDK1 = phosphoinositide-dependent kinase-1, TSC 1/2 = tuberous sclerosis complex 1/2, ATP = adenosine triphosphate, GDP = guanosine diphosphate, GTP = guanosine triphosphate, Rheb = Ras homolog enriched in brain, 4E-BP1 = eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1, S6K1 = ribosomal protein S6 kinase beta-1, rpS6 = ribosomal protein S6. The black blunt-ended lines indicate the main targets for therapeutic intervention. Created with BioRender.com.
Exploring mechanisms of constitutive PI3K/Akt/mTOR activation in AML identified mutations of RTKs (e.g., FLT3-ITD, c-KIT) or GTPases (KRAS, NRAS) as well as autocrine IGF-1/IGF-1R signaling responsible for dysregulation [111][112]. Aberrant PI3K/Akt/mTOR signaling activation is often associated with enhanced Akt phosphorylation, mediated by phosphorylation at S473 by PDK1 and T308 by mTORC2. The OS of AML patients presenting with Akt phosphorylation at these sites was found to be significantly shorter in several studies [105][108]. Furthermore, mTORC1 is activated in most AML patients, indicated by enhanced phosphorylation of its main downstream substrates 4E-BP1, S6K1, and rpS6 [23][113]. However, activation of mTORC1 and its downstream target may also occur independently of PI3K/Akt though parallel signaling pathways [114][115][116]. It is therefore important to dissect how PI3K/Akt/mTOR signaling converges with other signaling pathways, which may have clinical implications for selection of drugs targeting different signaling molecules.
About 30% of AML patients with normal karyotype present with an activating FLT3 receptor mutation, most often as FLT3-ITD, and is the major intercessory of PI3K/Akt/mTOR pathway dysregulation. FLT3 is a member of the class III RTK family and is important for the maintenance of hematopoietic homeostasis [117][118][119]. FLT3-ITD exhibits ligand-independent constitutive tyrosine kinase activity and activates signaling pathways including PI3K/Akt/mTOR [120]. However, regulatory p85 subunit of PI3K does not bind to the FLT3 receptor, nor is it tyrosine phosphorylated after FLT3 ligand stimulation. Instead, p85 associated with SH2 domain-containing protein tyrosine phosphatase-2 (SHP-2) and SHIP, in murine Ba/F3 cells stably transfected with human FLT3-ITD [121]. FLT3-ITD expression in Ba/F3 cells was associated with constitutive activation of Akt and concomitant phosphorylation of FoxO3a [112][122]. FoxO3a has an important role in apoptosis and cell cycle regulation [123]. FLT3-ITD was shown to constitutively activate Akt, and concomitantly phosphorylate FoxO3a, suppressing the expression of FoxO3a target genes encoding for p27 and pro-apoptotic Bcl-2 family member, Bim [112][124]. FLT3-ITD negatively regulates FoxO3a, thereby suppressing FoxO3a-mediated apoptosis and bypassing the G1 cell cycle blockade.
3.3. Targeting the PI3K/Akt/mTOR Signaling Pathway in AML
Preclinical evidence underlines the significant role of PI3K/Akt/mTOR signaling in leukemia initiation and maintenance. There is considerable interest targeting PI3K/Akt/mTOR signaling for AML treatment, which has resulted into the rapid development of small molecule compounds that target either a single or multiple kinase (Figure 3). PI3K-Targeting molecules can be divided into isoform-specific PI3K inhibitors and ATP-competitive pan-PI3K inhibitors. The PI3K p110δ catalytic subunit is consistently expressed at a high level in AML blasts, making it an attractive therapeutic target for AML [125]. Idelalisib (also referred to as CAL-101), for example, is a p110δ inhibitor that is currently under Phase 3 clinical investigation for the treatment of B-cell malignancies [126]. Treatment of AML cells with idelalisib inhibited ribosomal RNA (rRNA) synthesis and cell proliferation by suppressing Akt phosphorylation with a greater effect observed in cells expressing higher levels of p110δ [127]. Pan-PI3K inhibitors target all isoforms of PI3K and may exert broader anti-leukemic effects but at the expense of higher toxicity. mTOR inhibitors include both ATP-pocket and allosteric mTOR binding drugs, e.g., rapalogs, such as everolimus and temsirolimus. Both drugs derive from the natural macrolide rapamycin and act by associating with immunophillin FK506 binding protein 12 (FKBP12), which in turn binds and inhibits mTORC1, although after lengthy exposure they inhibit also mTORC2 [128]. mTOR inhibitors represent the first class of PI3K/Akt/mTOR-directed therapies and yielded promising anti-proliferative effects without inhibition of normal CD34+ cells in preclinical settings [129]. The anti-leukemic effects were associated with reduced phosphorylation of S6K1 and 4E-BP1 and could be enhanced in combination with conventional cytotoxic drugs [23]. An important drawback of inhibiting mTORC1 is the increased phosphorylation of Akt. Dual PI3K and mTOR inhibitors block both the upstream and downstream targets of Akt, thereby circumventing the increased PI3K and Akt signaling subsequent to mTORC1 inhibition [130]. Dual PI3K/mTOR inhibitor dactolisib efficiently blocked PI3K and mTORC1 signaling and mTORC2 activity [131]. Furthermore, dactolisib inhibited protein translation in AML cells, reducing cell growth and inducing apoptosis without affecting survival of normal CD34+ cells. A small number of Akt inhibitors have been developed, but they are rarely evaluated in preclinical or clinical settings as the development of Akt inhibitors has long been hampered by high structural similarity of the Akt catalytic domain to that of other kinases of the AGC kinase family (named after the representative protein kinase A, G, and C families) [91][132].
References
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Wang, F.; Loghavi, S.; Bueso-Ramos, C.; Gumbs, C.; Little, L.; Song, X.; Zhang, J.; Kadia, T.; Borthakur, G.; et al. Late relapse in acute myeloid leukemia (AML): Clonal evolution or therapy-related leukemia? Blood Cancer J. 2019, 9, 7. [Google Scholar] [CrossRef]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648.
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Guida, M.; Accordi, B.; Mediani, L.; Martelli, A.M.; Barozzi, P.; Petricoin, E.; Liotta, L.; Milani, G.; Giordan, M.; et al. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014, 28, 2197–2205.
- Hou, H.-A.; Tien, H.-F. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020, 27.
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A promising option for acute myeloid leukemia. Drug Des. Dev. Ther. 2019, 13, 1117–1125.
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef]
- Guzman, M.L.; Swiderski, C.F.; Howard, D.S.; Grimes, B.A.; Rossi, R.M.; Szilvassy, S.J.; Jordan, C.T. Preferential induction of apoptosis for primary human leukemic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16220–16225. [Google Scholar] [CrossRef]
- Zhou, J.; Ching, Y.Q.; Chng, W.-J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500.
- Li, F.; Sethi, G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Zhou, J.; Chooi, J.-Y.; Ching, Y.Q.; Quah, J.Y.; Toh, S.H.-M.; Ng, Y.; Tan, T.Z.; Chng, W.-J. NF-κB promotes the stem-like properties of leukemia cells by activation of LIN28B. World J. Stem Cells 2018, 10, 34–42.
- Birkenkamp, K.U.; Geugien, M.; Schepers, H.; Westra, J.; Lemmink, H.H.; Vellenga, E. Constitutive NF-kappaB DNA-binding activity in AML is frequently mediated by a Ras/PI3-K/PKB-dependent pathway. Leukemia 2004, 18, 103–112.
- Xu, Q.; Simpson, S.-E.; Scialla, T.J.; Bagg, A.; Carroll, M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003, 102, 972–980. [Google Scholar] [CrossRef]
- Xu, Q.; Thompson, J.E.; Carroll, M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 2005, 106, 4261–4268. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Frasson, C.; Chiarini, F.; D’Avella, D.; Accordi, B.; Anselmi, L.; Barozzi, P.; Forghieri, F.; Luppi, M.; Martelli, A.M.; et al. Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv. Biol. Regul. 2018, 68, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Grandage, V.L.; Gale, R.E.; Linch, D.C.; Khwaja, A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia 2005, 19, 586–594.
- Xu, Q.; Thompson, J.E.; Carroll, M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 2005, 106, 4261–4268.
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341.
- Vachhani, P.; Bose, P.; Rahmani, M.; Grant, S. Rational combination of dual PI3K/mTOR blockade and Bcl-2/-xL inhibition in AML. Physiol. Genom. 2014, 46, 448–456.
- Markham, A. Copanlisib: First Global Approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Phosphoinositide. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef]
- Miller, B.W.; Przepiorka, D.; de Claro, R.A.; Lee, K.; Nie, L.; Simpson, N.; Gudi, R.; Saber, H.; Shord, S.; Bullock, J.; et al. FDA approval: Idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin. Cancer Res. 2015, 21, 1525–1529.
- Nepstad, I.; Hatfield, K.J.; Gronningsaeter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907.
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156.
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vadas, O.; Burke, J.E.; Zhang, X.; Berndt, A.; Williams, R.L. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci. Signal. 2011, 4, re2.
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef]
- Brock, C.; Schaefer, M.; Reusch, H.P.; Czupalla, C.; Michalke, M.; Spicher, K.; Schultz, G.; Nürnberg, B. Roles of Gβγ in membrane recruitment and activation of p110γ/p101 phosphoinositide 3-kinase γ. J. Cell Biol. 2003, 160, 89–99.
- Kok, K.; Nock, G.E.; Verrall, E.A.G.; Mitchell, M.P.; Hommes, D.W.; Peppelenbosch, M.P.; Vanhaesebroeck, B. Regulation of p110delta PI 3-kinase gene expression. PLoS ONE 2009, 4, e5145.
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Kiyatkin, A.; Aksamitiene, E.; Markevich, N.I.; Borisov, N.M.; Hoek, J.B.; Kholodenko, B.N. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J. Biol. Chem. 2006, 281, 19925–19938.
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling Specificity in the Akt Pathway in Biology and Disease. Adv. Biol. Regul. 2014, 55, 28–38.
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594.
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522.
- Liu, Q.; Turner, K.M.; Alfred Yung, W.K.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, M.-C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42.
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stöckli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943.
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405.
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541.
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24.
- Hazen, A.L.; Smith, M.J.; Desponts, C.; Winter, O.; Moser, K.; Kerr, W.G. SHIP is required for a functional hematopoietic stem cell niche. Blood 2009, 113, 2924–2933.
- Luo, J.-M.; Liu, Z.-L.; Hao, H.-L.; Wang, F.-X.; Dong, Z.-R.; Ohno, R. Mutation analysis of SHIP gene in acute leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2004, 12, 420–426.
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Park, S.R.; Yoo, Y.J.; Ban, Y.-H.; Yoon, Y.J. Biosynthesis of rapamycin and its regulation: Past achievements and recent progress. J. Antibiot. 2010, 63, 434–441.
- Gibbons, J.J.; Abraham, R.T.; Yu, K. Mammalian target of rapamycin: Discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin. Oncol. 2009, 36 (Suppl. S3), S3–S17
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576.
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J. Biol. Chem. 2003, 278, 10189–10194.
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.W.Y.; Fryer, L.G.D.; Carling, D.; Shepherd, P.R. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J. Biol. Chem. 2004, 279, 15719–15722. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 2005, 280, 26089–26093.
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71.
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190.
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221.
- Hardie, D.G. New roles for the LKB1-->AMPK pathway. Curr. Opin. Cell Biol. 2005, 17, 167–173. [Google Scholar] [CrossRef]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428.
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590.
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328.
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494.
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577.
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437.
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127.
- Han, J.W.; Pearson, R.B.; Dennis, P.B.; Thomas, G. Rapamycin, wortmannin, and the methylxanthine SQ20006 inactivate p70s6k by inducing dephosphorylation of the same subset of sites. J. Biol. Chem. 1995, 270, 21396–21403. [Google Scholar] [CrossRef]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175.
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348.
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 2004, 24, 3112–3124.
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382.
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415.
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; Guan, K.-L.; Morrison, S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605.
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.-L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408.
- Tothova, Z.; Gilliland, D.G. FoxO transcription factors and stem cell homeostasis: Insights from the hematopoietic system. Cell Stem Cell 2007, 1, 140–152. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112.
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828.
- Fransecky, L.; Mochmann, L.H.; Baldus, C.D. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 2015, 3, 2.
- Gao, Y.; Gao, J.; Li, M.; Zheng, Y.; Wang, Y.; Zhang, H.; Wang, W.; Chu, Y.; Wang, X.; Xu, M.; et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J. Hematol. Oncol. 2016, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Kobayashi, M.; Ramdas, B.; Chatterjee, A.; Ma, P.; Mali, R.S.; Carlesso, N.; Liu, Y.; Plas, D.R.; Chan, R.J.; et al. S6K1 regulates hematopoietic stem cell self-renewal and leukemia maintenance. J. Clin. Investig. 2016, 126, 2621–2625. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Kapur, R. Role of mTORC1-S6K1 signaling pathway in regulation of hematopoietic stem cell and acute myeloid leukemia. Exp. Hematol. 2017, 50, 13–21.
- Gao, Y.; Gao, J.; Li, M.; Zheng, Y.; Wang, Y.; Zhang, H.; Wang, W.; Chu, Y.; Wang, X.; Xu, M.; et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J. Hematol. Oncol. 2016, 9, 36.
- Zabkiewicz, J.; Pearn, L.; Hills, R.K.; Morgan, R.G.; Tonks, A.; Burnett, A.K.; Darley, R.L. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica 2014, 99, 858–864.
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.
- Ghosh, J.; Kobayashi, M.; Ramdas, B.; Chatterjee, A.; Ma, P.; Mali, R.S.; Carlesso, N.; Liu, Y.; Plas, D.R.; Chan, R.J.; et al. S6K1 regulates hematopoietic stem cell self-renewal and leukemia maintenance. J. Clin. Investig. 2016, 126, 2621–2625.
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259.
- Zeng, Z.; Shi, Y.X.; Tsao, T.; Qiu, Y.; Kornblau, S.M.; Baggerly, K.A.; Liu, W.; Jessen, K.; Liu, Y.; Kantarjian, H.; et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012, 120, 2679–2689. [Google Scholar] [CrossRef]
- Reikvam, H.; Nepstad, I.; Bruserud, Ø.; Hatfield, K.J. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843.
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26.
- Gallay, N.; Dos Santos, C.; Cuzin, L.; Bousquet, M.; Simmonet Gouy, V.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Récher, C. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009, 23, 1029–1038.
- Bertacchini, J.; Frasson, C.; Chiarini, F.; D’Avella, D.; Accordi, B.; Anselmi, L.; Barozzi, P.; Forghieri, F.; Luppi, M.; Martelli, A.M.; et al. Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv. Biol. Regul. 2018, 68, 2–9.
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef]
- Min, Y.H.; Eom, J.I.; Cheong, J.W.; Maeng, H.O.; Kim, J.Y.; Jeung, H.K.; Lee, S.T.; Lee, M.H.; Hahn, J.S.; Ko, Y.W. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its significance as a prognostic variable. Leukemia 2003, 17, 995–997.
- Martelli, A.M.; Nyåkern, M.; Tabellini, G.; Bortul, R.; Tazzari, P.L.; Evangelisti, C.; Cocco, L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006, 20, 911–928. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Aasebø, E.; Hernandez-Valladares, M.; Brenner, A.K.; Bartaula-Brevik, S.; Berven, F.; Selheim, F.; Skavland, J.; Gjertsen, B.T.; et al. Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin. Ther. Targets 2018, 22, 639–653.
- Chapuis, N.; Tamburini, J.; Cornillet-Lefebvre, P.; Gillot, L.; Bardet, V.; Willems, L.; Park, S.; Green, A.S.; Ifrah, N.; Dreyfus, F.; et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: Therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010, 95, 415–423. [Google Scholar] [CrossRef]
- Brandts, C.H.; Sargin, B.; Rode, M.; Biermann, C.; Lindtner, B.; Schwäble, J.; Buerger, H.; Müller-Tidow, C.; Choudhary, C.; McMahon, M.; et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005, 65, 9643–9650.
- Tamburini, J.; Green, A.S.; Bardet, V.; Chapuis, N.; Park, S.; Willems, L.; Uzunov, M.; Ifrah, N.; Dreyfus, F.; Lacombe, C.; et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627.
- Tamburini, J.; Green, A.S.; Bardet, V.; Chapuis, N.; Park, S.; Willems, L.; Uzunov, M.; Ifrah, N.; Dreyfus, F.; Lacombe, C.; et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Minden, M.D.; Hedley, D.W. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp. Hematol. 2006, 34, 1183–1191.
- Chu, S.H.; Heiser, D.; Li, L.; Kaplan, I.; Collector, M.; Huso, D.; Sharkis, S.J.; Civin, C.; Small, D. FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell 2012, 11, 346–358. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Kikushige, Y.; Yoshimoto, G.; Miyamoto, T.; Iino, T.; Mori, Y.; Iwasaki, H.; Niiro, H.; Takenaka, K.; Nagafuji, K.; Harada, M.; et al. Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J. Immunol. 2008, 180, 7358–7367.
- Chen, W.; Drakos, E.; Grammatikakis, I.; Schlette, E.J.; Li, J.; Leventaki, V.; Staikou-Drakopoulou, E.; Patsouris, E.; Panayiotidis, P.; Medeiros, L.J.; et al. mTOR signaling is activated by FLT3 kinase and promotes survival of FLT3-mutated acute myeloid leukemia cells. Mol. Cancer 2010, 9, 292.
- Zhang, S.; Broxmeyer, H.E. p85 subunit of PI3 kinase does not bind to human Flt3 receptor, but associates with SHP2, SHIP, and a tyrosine-phosphorylated 100-kDa protein in Flt3 ligand-stimulated hematopoietic cells. Biochem. Biophys. Res. Commun. 1999, 254, 440–445.
- Scheijen, B.; Ngo, H.T.; Kang, H.; Griffin, J.D. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004, 23, 3338–3349.
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986.
- Santo, E.E.; Stroeken, P.; Sluis, P.V.; Koster, J.; Versteeg, R.; Westerhout, E.M. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer Res. 2013, 73, 2189–2198.
- Sujobert, P.; Bardet, V.; Cornillet-Lefebvre, P.; Hayflick, J.S.; Prie, N.; Verdier, F.; Vanhaesebroeck, B.; Muller, O.; Pesce, F.; Ifrah, N.; et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005, 106, 1063–1066.
- Gopal, A.; Graf, S. Idelalisib for the treatment of non-Hodgkin lymphoma. Expert Opin. Pharmacother. 2016, 17, 265–274.
- Nguyen, L.X.T.; Sesay, A.; Mitchell, B.S. Effect of CAL-101, a PI3Kδ inhibitor, on ribosomal rna synthesis and cell proliferation in acute myeloid leukemia cells. Blood Cancer J. 2014, 4, e228.
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822.
- Xu, Q.; Simpson, S.-E.; Scialla, T.J.; Bagg, A.; Carroll, M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003, 102, 972–980.
- Carneiro, B.A.; Kaplan, J.B.; Altman, J.K.; Giles, F.J.; Platanias, L.C. Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol. Ther. 2015, 16, 648–656.
- Chapuis, N.; Tamburini, J.; Green, A.S.; Vignon, C.; Bardet, V.; Neyret, A.; Pannetier, M.; Willems, L.; Park, S.; Macone, A.; et al. Dual Inhibition of PI3K and mTORC1/2 Signaling by NVP-BEZ235 as a New Therapeutic Strategy for Acute Myeloid Leukemia. Clin. Cancer Res. 2010, 16, 5424–5435.
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885.


