 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmed Sheriff | + 1916 word(s) | 1916 | 2020-09-14 08:49:12 | | | |
| 2 | Catherine Yang | Meta information modification | 1916 | 2020-09-17 07:45:37 | | |
Video Upload Options
Almost every kind of inflammation in the human body is accompanied by rising C-reactive protein (CRP) concentrations. This can include bacterial and viral infection, chronic inflammation and so-called sterile inflammation triggered by (internal) acute tissue injury. CRP is part of the ancient humoral immune response and secreted into the circulation by the liver upon respective stimuli. Its main immunological functions are the opsonization of biological particles (bacteria and dead or dying cells) for their clearance by macrophages and the activation of the classical complement pathway. This not only helps to eliminate pathogens and dead cells, which is very useful in any case, but unfortunately also to remove only slightly damaged or inactive human cells that may potentially regenerate with more CRP-free time. CRP action severely aggravates the extent of tissue damage during the acute phase response after an acute injury and therefore negatively affects clinical outcome. CRP is therefore a promising therapeutic target to rescue energy-deprived tissue either caused by ischemic injury (e.g., myocardial infarction and stroke) or by an overcompensating immune reaction occurring in acute inflammation (e.g., pancreatitis) or systemic inflammatory response syndrome (SIRS; e.g., after transplantation or surgery). Selective CRP apheresis can remove circulating CRP safely and efficiently. We explain the pathophysiological reasoning behind therapeutic CRP apheresis and summarize the broad span of indications in which its application could be beneficial with a focus on ischemic stroke as well as the results of this therapeutic approach after myocardial infarction.
1. General Introduction
Inflammatory processes involve a plethora of signaling pathways and affect the whole body, even if their origin is most often locally restricted in an acute setting. Mounting an inflammatory response is the body’s strategy to primarily eliminate any cause of tissue damage and subsequently repair the injury [1]. This is rooted in the evolutionary background that damage is mainly caused by pathogens or at least exacerbated by them within an external wound. In this case the elicited inflammation is beneficial in fighting infiltrating bacteria or viruses as well as restoring tissue homeostasis. However, healing of injured tissue often happens at the cost of still healthy tissue/cells and involves additional cell death as collateral damage [2]. In specific situations, these negative effects outweigh the positive aspects of the inflammatory reaction. Whenever an injury is “sterile”, meaning it occurred internally without pathogen involvement, inflammation aggravates deterioration by elimination of additional cells, which were either vital or only slightly and reversibly impaired. This happens for example after ischemic injury like stroke or myocardial infarction, leading to a larger extent of organ damage, increased scarring and thereby worsening clinical outcome [3][4]. Likewise, negative effects dominate in situations when the immune system produces an excessive general reaction that is not justified by the trigger [5]. For example, during acute pancreatitis, a systemic inflammatory response syndrome (SIRS), or an acute bacterial or viral infection (Sepsis) the inflammation might cause widespread tissue injury, which might result in multiple organ failure [6].
Although a plentitude of proteins is involved in inflammation, many of them are cytokines or modulators that do not actively participate in the elimination of pathogens or cells [1]. Several mediator proteins play a key role.
One of the acute-phase mediators directly involved in these pro-inflammatory processes is C-reactive protein (CRP) which was discovered by Tillett and Francis in 1930 [7]. CRP is well-established as one of the most reliable markers of inflammation, rising dramatically during any type of inflammation. It has been shown that CRP as an inflammatory mediator not only reflects tissue damage, but also aggravates the severity of damage and contributes causally to course and outcome of various diseases [8]. Therefore, CRP has to be regarded not only as a marker, but also as an active pro-inflammatory protein.
2. Role of CRP
CRP is a sensitive, reliable and early indicator of inflammation and infection. Evolutionarily highly conserved, this pentameric molecule is part of the ancient humoral immune response and involved in various immunological pathways as a key mediator [9][10]. It is predominantly synthesized and secreted into the blood circulation by hepatic cells as a response to trauma, inflammation, or infection. In these situations, the proinflammatory cytokines interleukin 6 (IL-6) and, to a lesser extent, interleukin 1β (IL-1β) as well as tumor necrosis factor α (TNFα) induce CRP expression on the transcriptional level [11][12][13][14]. Following an acute phase stimulus, serum CRP values increase up to levels a few thousand times higher than the normal (healthy) concentration of human CRP (0.05 to 3000 mg/L) [15][16]. The half-life in plasma is about 19 h [17][18].
After secretion, CRP efficiently detects and opsonizes bacteria upon their infiltration and initiates their phagocytosis by activation of complement [19][20]. This is probably its original purpose as one of the most ancient proteins within the humoral immune system.
However, CRP also detects and binds to endogenous cells [21][22]. Cells, which are either apoptotic, energy-depleted, or simply exposed to stressors like the acidic and often ischemic environment of inflammation display conformational and biochemical changes of their membrane [23]. One of these changes is the formation of lyso-phosphatidylcholine (LPC) by partial hydrolyzation of phosphatidylcholine (PC). To this end, one of its two fatty acid groups is removed by the secretory phospholipase A2 type IIa (sPLA2 IIa) [24][25]. This phospholipase is secreted and activated by inflammation (IL-6) and marks the beginning of detrimental destruction of still viable tissue after e.g., ischemia [26][27][28][29]. LPC is thereby accessible in the plasma membrane of dead, damaged, or inflamed cells. The CRP pentamer binds to LPC with high avidity in a so-called cooperative manner and subsequently mediates the elimination of these cells, similarly to infiltrating pathogens, by activating the classical complement pathway [30][31][32][33][34][35]. Complement C1q binds to CRP directly and mediates the binding of C2–C4 [36]. Thus, these cells are irreversibly marked for phagocytes which dispose the marked cells. Phagocytes in turn secrete IL-6 which induces the synthesis of additional CRP by the liver, subsequently amplifying the immune response. This way, more cells become marked by CRP (Figure 1).
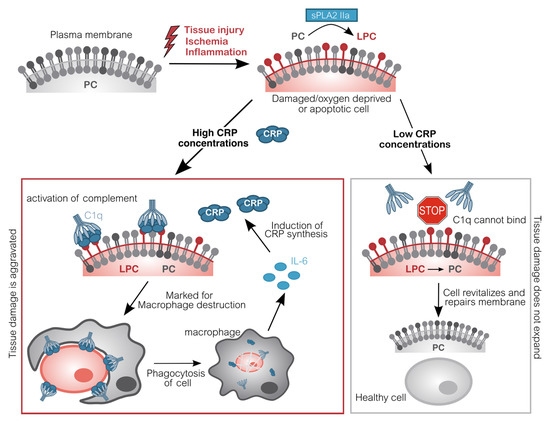
Figure 1. Molecular pathomechanism of CRP-mediated tissue damage. Upon inflammation or acute oxygen-deprivation, cells display a dramatic shortage of adenosine triphosphate (ATP). ATP is essential to prevent apoptosis which manifests in the outer cell membrane: Phosphatidylcholine (PC) is converted into lyso-phosphatidylcholine (LPC) by phospholipase (sPLA2 IIa). Due to the lack of ATP, this alteration cannot be reversed. CRP subsequently binds to LPC on anaerobic cells and recruits complement factors (C1q-C4). These opsonized cells will be disposed by phagocytes, which in turn induce CRP synthesis. Without CRP or in situations with low CRP concentrations (e.g., after CRP apheresis), energy deprived-cells are spared and may switch back to aerobic metabolism, repair molecular changes and revitalize again, leading to an overall reduced tissue damage [37][38][39][40]. CRP C-reactive protein; C1q Complement component 1q; IL-6 Interleukin 6; LPC Lysophosphatidylcholine; PC Phosphatidylcholine; sPLA2 IIa secretory phospholipase A2 type IIa.
Importantly, this mechanism facilitates binding of CRP to actually still vital cells, which may potentially regenerate with more CRP-free time. By interacting with complement, CRP triggers the destruction and therefore negatively affects the regeneration of tissue. By now, a large body of data obtained from animal experiments demonstrates that this CRP-mediated mechanism plays an active role in exacerbating ischemia and reperfusion-induced damage [41][42][43][44][37][45][38].
To date, no pharmacologic inhibitor of inflammation has been proven to be successful in ischemia-related injuries, since they all featured unfavorable pharmacokinetic profiles or serious side effects. Therefore, a different strategy is needed to target the detrimental inflammatory response [38][46]. Specifically, targeting avoidable organ damage caused by the action of CRP represents a promising therapeutic option [38][47]. Decreasing CRP levels could potentially protect salvageable cells and give them more time to recover. Therefore, removing CRP from the blood circulation interrupts the innate cascade and reduces tissue damage [39]. Accordingly, CRP apheresis may potentially present a promising, highly efficient, and well-tolerated therapeutic option.
3. CRP Apheresis
Extracorporeal apheresis refers to the physical removal of substances from the blood by means of filtration, precipitation or adsorption. Immunoadsorption defines the specific binding of an immunologic protein by an adsorber matrix. The elimination of pathogenic substances from the blood in extracorporeal apheresis constitutes an established therapeutic measure in the clinical routine of numerous diseases.
The CRP adsorber system (PentraSorb® CRP, Pentracor GmbH, Hennigsdorf, Germany) features an agarose-based resin, which contains a phosphocholine-derivative as ligand for CRP and is thereby capable of selectively depleting CRP from blood plasma with an efficiency of up to 94% (under laboratory conditions) [48]. CRP apheresis is executed in cycles, alternating between loading of the adsorber with plasma and regeneration of the column, that follows a fixed sequence of washing solutions. Loading and washing are controlled by a software module for automatic plasma flow management (ADAsorb, medicap clinic GmbH, Ulrichstein, Germany; Figure 2).
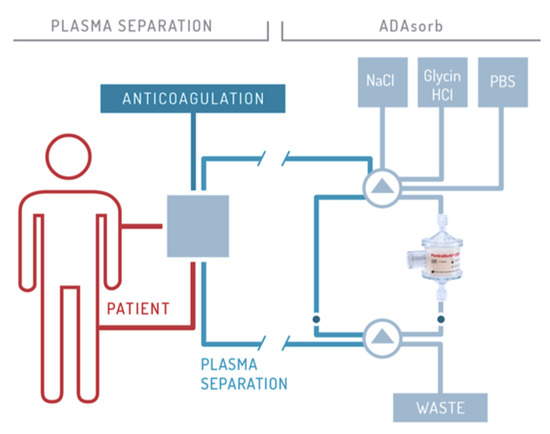
Figure 2. Schematic illustration of CRP apheresis. The procedure is described in detail by Ries et al. 2019 [40].
The main advantages of CRP apheresis are the selective removal of the agent by the highly specific ligand and the good controllability of the process, since unlimited plasma volumes can be processed to achieve the desired CRP reduction. Drugs are not removed by CRP apheresis. So far, no side-effects have been reported [40][49][50][51].
CRP Apheresis is a novel treatment option after ischemic tissue damage and has been investigated after acute myocardial infarction (CAMI-1 trial: "Selective depletion of C-reactive protein by therapeutic apheresis in acute myocardial infarction", DRKS ID: DRKS00008988). Just recenty, this multi-center clinical trials has been finished and first were shown in Case reports and a publication describing 13 patients as a preliminary report [39][40][49][50]. CRP apheresis will also be investigated after ischemic stroke (CASTRO-1 trial: "Selective Depletion of C-reactive Protein by Therapeutic Apheresis in Ischemic Stroke", ID: NCT0441728).
However, reduction of dramatically high CRP concentrations in other indications which are not defined by an ischemic pathophysiology could also be beneficial. The overcompensating immune reaction which often triggers SIRS after surgery, causes detrimental deterioration during acute pancreatitis, or mediates a cytokine storm after infection, could be dampened with CRP apheresis. Therefore, clinical trials investigating the safety and efficacy of CRP apheresis during pancreatitis and after coronary bypass surgery are ongoing (CAPRI1-study DRKS00014265; CABY1-study DRKS00013012).
4. Conclusions and Outlook
CRP has been established as a general biomarker of inflammation and infection in clinical practice. Recently, its role as a stable and highly useful prognostic factor for cardiovascular and cerebral disease in healthy individuals has been widely acknowledged and utilized [52][53]. However, the characterization of CRP as not only a biomarker but also a mediator or even trigger of immunological destruction of tissue is widely ignored [8][41][43].
Therapeutic CRP removal by immunoadsorption might present a logical and promising therapy for pathologies in which the extent of tissue damage is aggravated by inflammation and correlated with a worse clinical outcome, including ischemic events.
Preliminary evidence suggests that CRP apheresis induces very few side effects and features a low risk profile [40]. One drawback is that the procedure takes relatively long. Nevertheless, CRP apheresis fits well into the management of stroke patients because it does not collide with acute measures and may therefore complement methods aiming at reperfusion.
The acute inflammatory response has two facets. For one thing it plays a key role in initial host defense against infections. But on the downside, it can cause collateral damage of tissues. Especially in situations with an inciting sterile stimulus, the cost-benefit ratio is unfavorable.
CRP as an ancient protein of the innate immune system physiologically disposes cells and reacts to almost every disturbance of tissue homeostasis. Therefore, the span of potential indications for CRP apheresis is broad, and the ongoing clinical trials will illuminate whether this therapy is beneficial in these specific indications.
References
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, U.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831.
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525.
- Ong, S.-B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.-Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87.
- Anzai, T. Inflammatory Mechanisms of Cardiovascular Remodeling. Circ. J. 2018, 82, 629–635.
- Neher, M.D.; Weckbach, S.; Flierl, M.A.; Huber-Lang, M.; Stahel, P.F. Molecular mechanisms of inflammation and tissue injury after major trauma-is complement the “bad guy”? J. Biomed. Sci. 2011, 18, 90.
- Day, J.; Taylor, K. The systemic inflammatory response syndrome and cardiopulmonary bypass. Int. J. Surg. 2005, 3, 129–140.
- Tillett, W.S.; Francis, T. Serological Reactions in Pneumonia with a Non-Protein Somatic Fraction of Pneumococcus. J. Exp. Med. 1930, 52, 561–571.
- Kunze, R. C-Reactive Protein: From Biomarker to Trigger of Cell Death? Ther. Apher. Dial. 2019, 23, 494–496.
- Mortensen, R.F. C-Reactive Protein, Inflammation, and Innate Immunity. Immunol. Res. 2001, 24, 163–176.
- Du Clos, T.W. Pentraxins: Structure, Function, and Role in Inflammation. ISRN Inflamm. 2013, 2013, 1–22.
- Toniatti, C.; Arcone, R.; Majello, B.; Ganter, U.; Arpaia, G.; Ciliberto, G. Regulation of the human C-reactive protein gene, a major marker of inflammation and cancer. Mol. Biol. Med. 1990, 7, 199–212.
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 Participates in Transcriptional Activation of the C-reactive Protein Gene by Interleukin-6. J. Biol. Chem. 1996, 271, 9503–9509.
- Kramer, F.; Torzewski, J.; Kamenz, J.; Veit, K.; Hombach, V.; Dedio, J.; Ivashchenko, Y. Interleukin-1β stimulates acute phase response and C-reactive protein synthesis by inducing an NFκB- and C/EBPβ-dependent autocrine interleukin-6 loop. Mol. Immunol. 2008, 45, 2678–2689.
- Weinhold, B.; Bader, A.; Poli, V.; Rüther, U. Interleukin-6 is necessary, but not sufficient, for induction of the humanC-reactive protein gene in vivo. Biochem. J. 1997, 325, 617–621.
- Kushner, I.; Broder, M.L.; Karp, D. Control of the Acute Phase Response. J. Clin. Investig. 1978, 61, 235–242.
- Pietilä, K.; Harmoinen, A.; Hermens, W.; Simoons, M.L.; Van De Werf, F.; Verstraete, M. Serum C-reactive protein and infarct size in myocardial infarct patients with a closed versus an open infarct-related coronary artery after thrombolytic therapy. Eur. Hear. J. 1993, 14, 915–919.
- Dimitrijević, O.; Stojcevski, B.D.; Ignjatović, S.; Singh, N.M. Serial Measurements of C-Reactive Protein After Acute Myocardial Infarction in Predicting One-Year Outcome. Int. Hear. J. 2006, 47, 833–842.
- Gabriel, A.S.; Martinsson, A.; Wretlind, B.; Ahnve, S. IL-6 levels in acute and post myocardial infarction: Their relation to CRP levels, infarction size, left ventricular systolic function, and heart failure. Eur. J. Intern. Med. 2004, 15, 523–528.
- Szalai, A.J.; Briles, D.E.; Volanakis, J.E. Role of complement in C-reactive-protein-mediated protection of mice from Streptococcus pneumoniae. Infect. Immun. 1996, 64, 4850–4853.
- Mold, C.; Rodic-Polic, B.; Du Clos, T.W. Protection from Streptococcus pneumoniae Infection by C-Reactive Protein and Natural Antibody Requires Complement But Not Fcγ Receptors. J. Immunol. 2002, 168, 6375–6381.
- Chang, M.-K.; Binder, C.J.; Torzewski, M.; Witztum, J.L. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2002, 99, 13043–13048.
- Li, Y.P.; Mold, C.; Du Clos, T.W. Sublytic complement attack exposes C-reactive protein binding sites on cell membranes. J. Immunol. 1994, 152, 2995–3005.
- Sparkes, B.L.; Woods, K.; Roth, M.; Welti, R.; Fleming, S.D. Phospholipase A2 alters membrane lipid composition during ischemia/reperfusion (39.55). J. Immunol. 2009, 182, 39.55.
- Yagami, T.; Yamamoto, Y.; Koma, H. The Role of Secretory Phospholipase A2 in the Central Nervous System and Neurological Diseases. Mol. Neurobiol. 2013, 49, 863–876.
- Murakami, M.; Taketomi, Y.; Sato, H.; Yamamoto, K. Secreted phospholipase A2 revisited. J. Biochem. 2011, 150, 233–255.
- Fujioka, D.; Kawabata, K.-I.; Ishimoto, Y.; Suzuki, N.; Hanasaki, K.; Sato, R.; Hasebe, H.; Kobayashi, T.; Saito, Y.; Kanazawa, M.; et al. Abstract 1435: Reduction in Myocardial Ischemia-reperfusion Injury in Group X Secretory Phospholipase A2-deficient Mice. Circulation 2006, 114, II_275.
- Yano, T.; Fujioka, D.; Saito, Y.; Kobayashi, T.; Nakamura, T.; Obata, J.-E.; Kawabata, K.; Watanabe, K.; Watanabe, Y.; Mishina, H.; et al. Group V secretory phospholipase A2 plays a pathogenic role in myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2010, 90, 335–343.
- Nijmeijer, R.; Lagrand, W.K.; Baidoshvili, A.; Lubbers, Y.T.P.; Hermens, W.T.; Meijer, C.J.L.M.; Visser, C.A.; Hack, C.E.; Niessen, H.W.M. Secretory type II phospholipase A(2) binds to ischemic myocardium during myocardial infarction in humans. Cardiovasc. Res. 2002, 53, 138–146.
- Nijmeijer, R.; Willemsen, M.; Meijer, C.J.L.M.; Visser, C.A.; Verheijen, R.H.; Gottlieb, R.A.; Hack, C.E.; Niessen, H.W.M. Type II secretory phospholipase A2 binds to ischemic flip-flopped cardiomyocytes and subsequently induces cell death. Am. J. Physiol. Circ. Physiol. 2003, 285, H2218–H2224.
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754.
- Goda, T.; Miyahara, Y. Calcium-independent binding of human C-reactive protein to lysophosphatidylcholine in supported planar phospholipid monolayers. Acta Biomater. 2017, 48, 206–214.
- Kushner, I.; Kaplan, M.H. Studies of acute phase protein: I. An Immunohistochemical method for the localization of cx-reactive protein in rabbits. Association with necrosis in local inflammatory lesions. J. Exp. Med. 1961, 114, 961–974.
- Narkates, A.J.; Volanakis, J.E. C-reactive protein binding specificities: Artificial and natural phospholipid bilayers. Ann. N. Y. Acad. Sci. 1982, 389, 172–182.
- Kushner, I.; Rakita, L.; Kaplan, M.H. Studies of acute-phase protein. II. Localization of Cx-reactive protein in heart in induced myocardial infarction in rabbits. J. Clin. Investig. 1963, 42, 286–292.
- Vogt, B.; Führnrohr, B.; Müller, R.; Sheriff, A. CRP and the disposal of dying cells: Consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity 2007, 40, 295–298.
- Gaboriaud, C.; Juanhuix, J.; Gruez, A.; Lacroix, M.; Darnault, C.; Pignol, D.; Verger, D.; Fontecilla-Camps, J.C.; Arlaud, G.J. The Crystal Structure of the Globular Head of Complement Protein C1q Provides a Basis for Its Versatile Recognition Properties. J. Biol. Chem. 2003, 278, 46974–46982.
- Sheriff, A.; Schindler, R.; Vogt, B.; Abdel-Aty, H.; Unger, J.K.; Bock, C.; Gebauer, F.; Slagman, A.; Jerichow, T.; Mans, D.; et al. Selective apheresis of C-reactive protein: A new therapeutic option in myocardial infarction? J. Clin. Apher. 2014, 30, 15–21.
- Pepys, M.B.; Hirschfield, G.M.; Tennent, G.A.; Gallimore, J.R.; Kahan, M.C.; Bellotti, V.; Hawkins, P.N.; Myers, R.M.; Smith, M.D.; Polara, A.; et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 2006, 440, 1217–1221.
- Ries, W.; Heigl, F.; Garlichs, C.; Sheriff, A.; Torzewski, J. Die CRP-Apherese: Eine neue Therapiemöglichkeit bei Inflammation; Nephro-News, Medicom VerlagsgmbH Bruck: Mur, Austria, 2019; pp. 23–27.
- Ries, W.; Heigl, F.; Garlichs, C.; Sheriff, A.; Torzewski, J. Selective C-Reactive Protein-Apheresis in Patients. Ther. Apher. Dial. 2019, 23, 570–574.
- Gill, R.; Kemp, J.A.; Sabin, C.; Pepys, M.B. Human C-Reactive Protein Increases Cerebral Infarct Size after Middle Cerebral Artery Occlusion in Adult Rats. Br. J. Pharmacol. 2004, 24, 1214–1218.
- Hack, C.; Wolbink, G.-J.; Schalkwijk, C.; Speijer, H.; Hermens, W.T.; Bosch, H.V.D. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol. Today 1997, 18, 111–115.
- Griselli, M.; Herbert, J.; Hutchinson, W.; Taylor, K.; Sohail, M.; Krausz, T.; Pepys, M.B. C-Reactive Protein and Complement Are Important Mediators of Tissue Damage in Acute Myocardial Infarction. J. Exp. Med. 1999, 190, 1733–1740.
- Nijmeijer, R.; Lagrand, W.K.; Lubbers, Y.T.P.; Visser, C.A.; Meijer, C.J.L.M.; Niessen, H.W.M.; Hack, C.E. C-Reactive Protein Activates Complement in Infarcted Human Myocardium. Am. J. Pathol. 2003, 163, 269–275.
- Slagman, A.; Bock, C.; Abdel-Aty, H.; Vogt, B.; Gebauer, F.; Janelt, G.; Wohlgemuth, F.; Morgenstern, R.; Yapici, G.; Puppe, A.; et al. Specific Removal of C-Reactive Protein by Apheresis in a Porcine Cardiac Infarction Model. Blood Purif. 2011, 31, 9–17.
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47.
- Prasad, K. C-Reactive Protein (CRP)-Lowering Agents. Cardiovasc. Drug Rev. 2006, 24, 33–50.
- Mattecka, S.; Brunner, P.; Hähnel, B.; Kunze, R.; Vogt, B.; Sheriff, A. PentraSorb C-Reactive Protein: Characterization of the Selective C-Reactive Protein Adsorber Resin. Ther. Apher. Dial. 2019, 23, 474–481.
- Boljevic, D.; Nikolic, A.; Rusovic, S.; Lakcevic, J.; Bojic, M.; Balint, B. A Promising Innovative Treatment for ST-Elevation Myocardial Infarction: The Use of C-Reactive Protein Selective Apheresis: Case Report. Blood Purif. 2020, 1–5.
- Ries, W.; Sheriff, A.; Heigl, F.; Zimmermann, O.; Garlichs, C.D.; Torzewski, J. “First in Man”: Case Report of Selective C-Reactive Protein Apheresis in a Patient with Acute ST Segment Elevation Myocardial Infarction. Case Rep. Cardiol. 2018, 2018, 1–4.
- Torzewski, J.; Heigl, F.; Zimmermann, O.; Wagner, F.; Schumann, C.; Hettich, R.; Bock, C.; Kayser, S.; Sheriff, A. First-in-Man: Case Report of Selective C-Reactive Protein Apheresis in a Patient with SARS-CoV-2 Infection. Am. J. Case Rep. 2020, 21, e925020.
- Di Napoli, M.; Schwaninger, M.; Cappelli, R.; Ceccarelli, E.; Di Gianfilippo, G.; Donati, C.; Emsley, H.; Forconi, S.; Hopkins, S.J.; Masotti, L.; et al. Evaluation of C-Reactive Protein Measurement for Assessing the Risk and Prognosis in Ischemic Stroke. Stroke 2005, 36, 1316–1329.
- Peters, S.A.E.; Visseren, F.L.; Grobbee, D.E. Screening for C-reactive protein in CVD prediction. Nat. Rev. Cardiol. 2012, 10, 12–14.



