 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Javier Molina-Cerrillo | + 1638 word(s) | 1638 | 2020-09-11 07:37:25 | | | |
| 2 | Bruce Ren | Meta information modification | 1638 | 2020-09-14 03:18:22 | | |
Video Upload Options
Colon cancer is one of the most frequently diagnosed malignancies in adults, considering both its incidence and prevalence. Anatomically, the right colon is considered as being from the cecum to the splenic flexure, and the left colon is from the splenic flexure to the rectum. Sidedness is a surrogate of a wide spectrum of colorectal cancer (CRC) biology features (embryology, microbiome, methylation, microsatellite instability (MSI), BRAF, aging, KRAS, consensus molecular subtypes (CMS), etc.), which result in prognostic factors. Different molecular subtypes have been identified, according to genomic and transcriptomic criteria. A subgroup harboring a BRAF mutation has been described, and represents approximately 10% of the patients diagnosed with colon cancer. This subgroup has morphological, clinical, and therapeutic characteristics that differ substantially from patients who do not carry this genetic alteration. Unfortunately, there is no established standard of care for this particular cohort of patients.
1. Introduction
Colorectal cancer (CRC) incidence and mortality rates vary noticeably around the world. Overall, it is the third largest cause of cancer death, and the third most frequently diagnosed tumor, involving 11% of all new cases of cancer worldwide [1]. Although global mortality is decreasing, the subgroup of younger patients (< 50 years old) has experienced a growing incidence and mortality rate. Up to one quarter of patients have metastatic colorectal cancer (mCRC) disease at initial diagnosis, and nearly half of those who are candidates for primary tumor surgery will eventually develop metastasis, resulting in a 5-year survival rate of 14% [2].
Rates of CRC are substantially higher among males in comparison with females. In men, it entails the third most common type of cancer diagnosed, and the fourth most common cause of cancer death. Among females, it is the second most commonly diagnosed malignancy, and the third most common cause of cancer death [3].
The highest incidence rates are found in Australia, New Zealand, Europe, and North America, whereas South-Central Asia and Africa report a much lower incidence. In most Western countries, CRC incidence has been stable, or has experienced a slight increase within the last few years. In contrast, areas with a historical low incidence rate of CRC (Spain, Eastern Europe, or Eastern Asia) have experienced a rapid increase over the past few decades [3].
Up to 70% of CRC are sporadic and mainly associated with environmental and dietary factors. Less than 10% of patients with CRC have an inherited predisposition due to several genetic alterations, some of which can be already identified by specific tests. This category is subdivided into polyposis diseases (MUTYH-associated polyposis, familiar adenomatous polyposis, Cowden syndrome, and Peutz-Jeghers syndrome) and non-polyposis diseases, such as Lynch syndrome. Another pattern of presentation is the familial CRC that accounts for up to 25% of all CRC. This subset of patients has an increased risk of developing CRC, without fulfilling the criteria of the above-mentioned syndromes.
2. New Strategies
2.1. Tyrosine Kinase Inhibitors
When first line treatment fails in these patients, diverse strategies have been developed. Even though not many BRAF-mutant patients are fit to receive further treatment, this mutation is not associated with inferior outcomes after first line therapy. The use of second generation BRAF inhibitors in combination regimens are likely to work better than monotherapy.
Since BRAF-mutant cancer cells are highly dependent on MEK/ERK signaling, the combination of a BRAF inhibitor and a MEK inhibitor (double therapy) has shown a slight increased activity in comparison with either agent alone. Corcoran et al. demonstrated a reduced level of phosphorylated ERK in biopsies performed during a study of BRAFV600E-mutant mCRC treated with dabrafenib (a BRAF inhibitor) plus trametinib (a MEK inhibitor); 43 patients were enrolled with a 56% rate of stable disease, 12% partial response, and 2% complete response for more than 3 years [4]. BRAF inhibition downregulates the negative feedback signals from ERK, resulting in the activation of the EGFR pathway. This may explain the limited action of BRAF inhibitor in monotherapy in BRAF-mutant tumors, and would suggest that concomitant EGFR inhibition may overcome this resistance [5]. However, in the VE-BASKET study, the combination of vemurafenib (BRAF inhibitor) and cetuximab showed an ORR of 15% in 26 patients [6]. Other combinations under research have also demonstrated limited activity, suggesting that BRAFV600E inhibitors reactivate the EGFR signaling pathway [7]. A mechanism of resistance that is currently being evaluated converge on the formation of RAF dimers. Current RAF inhibitors block RAF monomers but not dimers. It is expected that third generation RAF inhibitors, which do inhibit RAF dimers, may shed light to the BRAF mutated mCRC management[8].
The combination of targeted therapies against EGFR and BRAF has reported, in a phase I trial, a partial response in 19 of 91 patients included (21%) and stable disease in 59 of 91 patients (65%), with an overall tumor growth control of 86%. The BEACON trial with 665 BRAF V600E mutated mCRC patients randomized in a 3-arm phase III trial to triplet therapy with encorafenib (BRAF inhibitor) plus binimetinib (MEK inhibitor) and cetuximab vs. encorafenib, plus cetuximab vs. a control arm (irinotecan/FOLFIRI + cetuximab) in patients RAS wt in a second or third line setting. The final results in the triplet combination confirmed ORR and median OS of 26% and 9.0 months, respectively, and 2% and 5.4 months, respectively, in the control arm. The doublet therapy showed a median OS of 8.4. adverse events at G3 or higher were 58%, 50%, and 61% in the triplet-, doublet- and control-arm group, respectively [9][10].
The knowledge about acquired mechanisms of resistance is not completely understood, and some possible explanations have been suggested. For example, activations of the PI3K/AKT pathway have been described in patients receiving BRAF inhibitors, in order to keep intracellular signaling via ERK [11]. Furthermore, overexpression of the hepatocyte growth factor (HGF), or its receptor c-MET, may lead resistance to BRAF inhibitors through the PI3K/AKT pathway [12]. However, this is not the only crosstalk pathway. From in vitro models, it has been described that BRAF inhibitors resulted in a feedback activation of EGFR signaling, in order to maintain ERK phosphorylation, suggesting it may be a new possible resistance pathway [13]. Additionally, the expression of aberrant spliced forms of BRAF V600E, such as BRAF V600E ΔEx, which are not sensitive to BRAF inhibitors, or truncated isoforms that continuously activate MEK/ERK signaling through dimerization, may also be involved in the resistance to BRAF inhibitors [14][15].
Moreover, resistance mechanisms due to complex genomic alterations have been described. BRAF amplifications are related to acquired resistance to BRAF and MEK inhibitors. It also seems that high BRAF mutant allele frequency may be related with high risk of primary disease refractoriness. [16] KRAS G13D amplifications have been also described in in vitro models of melanoma cells treated with MEK inhibitors [17].
The loss of Neurofibromin 1 (NF1), as a tumor suppressor that inhibits RAS, or the amplification of Cyclin D1, as a key factor in cell cycle regulation, were described in in vitro models as resistance pathways to BRAF inhibition .
2.2. Immunotherapy
BRAF mutations do not modify the response of MSI patients to immunotherapy. In general, 3–6% of stage IV CRC have a deficiency in DNA mismatch repair enzymes. Since pembrolizumab and nivolumab have shown activity against MSI mCRC, this may be a potential therapeutic option . A study evaluating the role of nivolumab in monotherapy in 74 MSI patients showed an ORR of 25% in BRAF-mutant tumors, 27% in KRAS mutated, and 41% if both were wild type . A combination of nivolumab with ipilimumab obtained similar results [18]. No randomized trials have been performed to compare dual checkpoint inhibitor therapy with monotherapy. However, indirect comparisons from the CheckMate 142 trial suggest that the combination may be superior. Nivolumab, pembrolizumab, and the combination of nivolumab-ipilimumab are approved by the FDA for patients with MSI or dMMR mCRC, which have progressed to other treatments [18].
3. Future Promising Strategies: Chemokine Receptors
Therapeutic research in the field of BRAF-mutant CRC is currently focused on new potential targets and combinations of known targeted therapies, with the aim of overcoming MAPK pathway resistance.
BRAF-mutant MSI CRC is related to the overexpression of stromal cell-derived factor-1 (SDF-1, also called CXCL12) and chemokine (C-X-C motif) receptor 4 (CXCR4), also known as fusin or CD184. CXCR4 is expressed in several cells from different organs, including colon, lung, liver, brain, and hematopoietic and progenitor cells, among others. CXCR4 belongs to a superfamily of G protein-couple receptors, and is functionally expressed in different types of cancer cells, including colorectal cancer cells. After its activation, it dissociates in two subunits: Gα, involved in regulating RAS/RAF and Gβγ, which activates PI3K/Akt/mTOR. There is a crosstalk between the EGFR and CXCL12/CXCR4 signaling pathways. CXCR4 is able to directly upregulate EGFR phosphorylation after its activation, but also indirectly, by increasing ERK phosphorylation [19][20][21][22] (Figure 1).
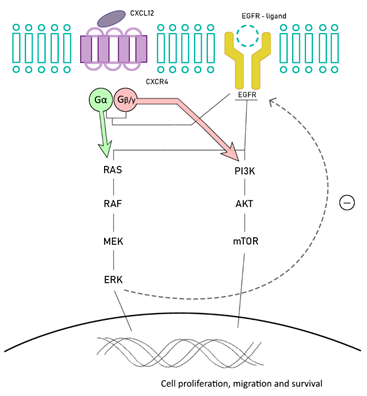
Figure 1. Chemokine (C-X-C motif) receptor 4 (CXCR4)/EGFR pathways crosstalk. CXCR4 is able to upregulate EGFR activation and finally ERK phosphorylation.
The CXCL12-CXCR4 axis regulates, among others, the migration and homing of lymphocytes to secondary lymphoid tissue and, also, for hematopoietic stem cells to the bone marrow. In CRC, this axis has demonstrated its role in promoting the migration, invasion (through angiogenesis), and transition of epithelial-mesenchymal tissue into neoplastic cells [19][20].
In a recent study of 78 primary CRC, CXCR4 expression was correlated with grading and response to first line chemotherapy, representing a strong and independent prognostic factor, since its high expression was correlated with a poor response in first line treatment, especially if anti-EGFR therapy was administered. A crosstalk between CXCR4 and VEGFR has been postulated by a synergistic activity from both ligands (CXCL12 and VEGF), which copes with the action of bevacizumab by restoring the angiogenesis. In fact, the high expression of CXCR4 and poor response to anti-EGFR was proven. Hence, inhibiting this axis may be a powerful strategy to deal with this resistance[22].
High CXCR4 expression is correlated with a poor histological differentiation, also being related to an increased risk of local recurrence and/or lymph node and distant metastasis in earlier stages, as well as worse OS (23 months vs. 9 months in tumors with low CXCR4 expression) [23].
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Przeglad Gastroenterologiczny 2019, 14, 89–103.
- Gómez-España, M.A.; Gallego, J.; González-Flores, E.; Maurel, J.; Páez, D.; Sastre, J.; Aparicio, J.; Benavides, M.; Feliu, J.; Vera, R. SEOM clinical guidelines for diagnosis and treatment of metastatic colorectal cancer (2018). Clin. Transl. Oncol. 2019, 21, 46–54.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-Mutant colorectal cancer. J. Clin. Oncol. 2015,33,4023–4031
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.;Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103.
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J., Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736.
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320
- Yaeger, R.; Yao, Z.; Hyman, D.M.; Hechtman, J.F.; Vakiani, E.; Zhao, H.; Su, W.; Wang, L.; Joelson, A.; Cercek, A.; et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res. 2017, 77, 6513–6523.
- Van Cutsem, E.; Cuyle, P.; Huijberts, S.; Schellens, J.; Elez, E.; Yaeger, R.; Fakih, M.; Montagut, C.; Peeters, M.; Desai, J.; et al. BEACON CRC study safety lead-in: Assessment of the BRAF inhibitor encorafenib + MEK inhibitor binimetinib + anti-epidermal growth factor receptor antibody cetuximab for BRAFV600E metastatic colorectal cancer. Ann. Oncol. 2018, 29 (Suppl. 5), V109.
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAFV600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643.
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R.; Dayyani, F., Jr.; Gopal,Y. N.; Jiang, Z.Q.; Wistuba, I.I.; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667.
- Spagnolo, F.; Ghiorzo, P.; Queirolo, P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014, 5, 10206–10221.
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual EGFR and BRAF blockade overcomes resistance to vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell Int. 2017, 17, 86.
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390.
- Vido, M.J.; Le, K., Hartsough, E.J.; Aplin, A.E. BRAF Splice Variant Resistance to RAF Inhibitor Requires Enhanced MEK Association. Cell Rep. 2018, 25, 1501–1510.
- Stagni, C.; Zamuner, C.; Elefanti, L.; Zanin, T.; Bianco, P.D.; Sommariva, A.; Fabozzi, A.; Pigozzo, J.; Mocellin, S.; Montesco, M.C.; et al. BRAF Gene Copy Number and Mutant Allele Frequency Correlate With Time to Progression in Metastatic Melanoma Patients Treated With MAPK Inhibitors. Mol. Cancer Ther. 2018, 17, 1332–1340.
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 Inhibitor Withdrawal Reverses Acquired Resistance Driven by BRAF V600E Amplification Whereas KRAS G13D Amplification Promotes EMT-chemoresistance. Nat. Commun. 2019, 10, 2030.
- Overman, M.J.; Lonardi, S.; Wong, K.Y.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779.
- Xu, C.; Zheng, L.; Li, D.; Chen, G.; Gu, J.; Chen, J.; Yao, Q. CXCR4 overexpression is correlated with poor prognosis in colorectal cancer. Life Sci. 2018, 208, 333–340.
- Yu, X.; Wang, D.; Wang, X.; Sun, S.; Zhang, Y.; Wang, S.; Miao, R., Xu, X., Qu, X. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J. Exp. Clin. Cancer Res. 2019, 38, 32.
- Gala, M.K.; Austin, T.; Ogino, S.; Chan, A.T. TFF2-CXCR4 axis is associated with BRAF V600E colon cancer. Cancer Prev. Res. 2015, 8, 614–619
- Ottaiano, A.; Scala, S.; Normanno, N.; Botti, G.; Tatangelo, F.; Di Mauro, A.; Capozzi, M.; Facchini, S.; Tafuto, S.; Nasti, G. Prognostic and Predictive Role of CXC Chemokine Receptor 4 in Metastatic Colorectal Cancer Patients. Appl. Immunohistochem. Mol. Morphol. 2020, doi:10.1097/PAI.0000000000000828.
- Kim, J.; Takeuchi, H.; Lam, S.T.; Turner, R.R.; Wang, H.J.; Kuo, C.; Foshag, L.; Bilchik, A. J.; Hoon, D.S. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J. Clin. Oncol. 2005, 23, 2744–2753.


