 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alicia Roque | + 3404 word(s) | 3404 | 2020-08-19 04:44:51 | | | |
| 2 | Bruce Ren | Meta information modification | 3404 | 2020-08-26 11:13:21 | | |
Video Upload Options
Protein molecules can be further modified after translation. Post-translational modifications (PTMs) are responsible for most of the proteome diversity and often modulate critical protein functions in health and disease. Histone H1 is a chromatin structural protein, which contains many PTMs that may alter chromatin structure and function. In this entry, we present a summary of the post-translational modifications (PTMs) identified, up to date, in histone H1 from lower eukaryotes to humans. We also highlight the use of different proteomic strategies, as well as the technical challenges involved in mapping H1 PTMs.
1. Introduction
Histone H1 is a key chromatin structural protein, which mediates higher-order chromatin folding. H1 is also emerging as an important epigenetic mark and regulator of gene expression and cellular differentiation. Metazoan H1 has three structural domains: a short N-terminal domain (NTD), a central globular domain (GD), and a long C-terminal domain (CTD). The globular domain contains a winged-helix motif and is responsible for the binding of H1 to nucleosomal DNA. Both terminal domains are intrinsically disordered and have a positive net charge in physiological conditions due to the abundance of lysine residues [1][2]. The CTD is considered the main determinant of H1-driven chromatin compaction [3]. This domain is also responsible for the preference for the scaffold-associated regions (SAR-DNA) [4], for the interaction with apoptotic nuclease DFF40 [5], with the β-amyloid peptide, and the formation of amyloid-like fibers [6].
Histone H1, also known as linker histone, is the most divergent and heterogeneous group of histones. It has been suggested that the first H1-like proteins may have appeared early in evolution in eubacteria, while the sequence winged-helix motif, present in the GD, appeared much later, in protists [7]. Detection of putative recombination points suggest that this process may have been involved in the acquisition of the H1 tripartite structure [8]. In lower eukaryotes, histone H1 is very heterogeneous. Some protists, like Tetrahymena thermophila, are only lysine-rich with a sequence composition similar to bacterial H1-like proteins and the CTD of metazoans, while the Saccharomyces cerevisiae version of H1, HhoI, contains a second winged-helix motif at its C-terminal end [9]. However, it has been reported more recently that the high mobility group protein HMO1 functions as a linker histone in Saccharomyces cerevisiae [10].
In mammals, 11 H1 subtypes have been identified [11]. Subtypes H1.0–H1.5 and H1x are differentially expressed in somatic cells, while subtypes H1t, HILS1, and H1T2 are expressed in male germinal cells, and H1oo is expressed in oocytes [12]. Orthologous genes are readily identified across mammalian species, but they are rarely detected in moderately distant phyla. Mammalian H1 subtypes differ in their evolution velocities [13], expression patterns [12], chromatin binding affinity [14][15][16], and genomic distribution [17][18][19][20], among other features. The presence of post-translational modifications (PTMs) adds a new level of complexity to H1 diversity, as individual PTMs or their interplay can modulate protein structure and function. In this entry, we present a comprehensive summary of the PTMs identified in H1, from lower eukaryotes to humans.
2. Analysis of histone H1 PTMs.
The first post-translational modification in H1 was described in the 70s, when phosphorylated H1 was identified in different species, throughout the tree of life. In a relatively short period, H1 phosphorylation was reported in protists such as Physarum polycephalum [21] and Tetrahymena sp.[22][23], and also in animals including Drosophila melanogaster [24], chicken erythrocytes [25], mammalian cell lines (rat hepatoma cells (HTC), and Chinese hamster ovary cells (CHO)) [26][27]. The identification of phosphorylation was carried out based on the changes in H1 electrophoretic mobility and on the incorporation of 32P and/or phosphatase treatment. At the time, mapping the phosphate groups to individual residues was still elusive, and significant amounts of the modified protein were necessary for its detection.
The development of mass spectrometry-based proteomics has represented a significant breakthrough in the identification of post-translational modifications, allowing mapping of PTMs to specific residues as well as the detection of low abundant modified species. There are three main types of proteomic strategies, depending on the molecule analyzed by mass spectrometry: top-down, middle-down, and bottom-up. Top-down proteomics analyzes intact proteins, while middle-down and bottom-up proteomics analyze large protein fragments/domains and small peptides, respectively [28]. PTM mapping and identification is mostly carried out by bottom-up proteomics. In this type of proteomic strategy, intact proteins are digested into small peptides, which are analyzed by tandem mass spectrometry (MS/MS). During MS/MS, ionized peptides are selected in the first mass analyzer, fragmented, and the m/z of the product ions is determined in the second mass analyzer. Mass differences between product ions allow for peptide sequencing and PTM assignment. The availability of mass analyzers of high mass accuracy has allowed for the distinction between quasi-isobaric PTMs, trimethylation/acetylation, and dimethylation/formylation [29][30]
Analysis of histone H1 PTMs by bottom-up proteomics has represented a technical challenge for two main reasons. First, H1 amino acid composition is characterized by a high content of lysine residues. Therefore, digestion with trypsin yields peptides that are relatively small and hydrophilic, which are difficult to detect by MS due to poor retention in the C18 Reverse-Phase Ion-Pairing High Performance Liquid Chromatography (RP-HPLC) column. As a result, regions with the highest density of lysine residues such as the CTD tend to have low coverage. This problem has been addressed by propionylation of amine groups in the protein (N-terminal amines, and free and monomethylated lysine ε-amino groups) before or after tryptic digestion [31][32]. This modification has significantly improved the coverage of H1 by MS.
The second challenging aspect is the presence in variable quantities of multiple subtypes or variants in higher eukaryotes. In particular, mammalian subtypes H1.1–H1.5 have more than 60% of sequence identity, and the sequence of the GD is almost identical [33]. Thus, many promiscuous peptides, matching the sequence of several subtypes, are often found in bottom-up studies. Several strategies have been used for the assignment of subtype-specific PTMs. Separation of individual subtypes before proteolytic digestion have been performed by capillary electrophoresis [34] and by 2D-electrophoresis. The latter used acetic-urea (AU) or triton-acetic-urea (TAU) electrophoresis in the first dimension, and Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis (SDS-PAGE) in the second dimension [35][36][37]. Combining bottom-up and top-down proteomics has also been used to assign PTMs to individual subtypes[38][39][40][41][42].
Bottom-up proteomics has identified and mapped a wide variety of PTMs in several eukaryotes (summarized in Tables S1–S3). To our knowledge, at least 13 PTM types have been identified in H1 including phosphorylation, methylation, acetylation, citrullination, crotonylation, ubiquitylation, formylation, 2-hydroxyisobutyrylation, and ADP-ribosylation (parylation) [42][43][44][45]. In this section, a brief overview of the PTMs identified in several model organisms is made. For consistency, in all cases, the modified residue number corresponds to the mature protein, which lacks the initial methionine. Therefore, for some modifications, the position number is not the same as the number in the original reference.
2.1 Histone H1 PTMs identified in Tetrahymenta thermophila, Saccharomyces cerevisiae, and Drosophila melanogaster.
Phosphorylation of histone H1 in Tetrahymena thermophila was detected during the 70s. However, the complete characterization of phosphorylation of macronuclear H1 by mass spectrometry, in vegetative growing cells and starved cells, was not performed until 2006 (Figure 1, Table S1). This study confirmed the five phosphosites previously identified by Edman-sequencing and peptide microsequencing [46][47], which included three consensus cyclin-dependent kinases (CDK) sites with the sequence S/T-P-X-K/R: T34, T46, and T53, and two non-canonical CDK sites: S4 and S5. Additionally, the analysis identified two novel phosphorylation sites, S42 and S44, and two novel acetylation sites, K77/78 and K154 (Figure 1, Table S1).
Separation of phosphorylated species from unphosphorylated up to heptaphosphorylated H1, by cation-exchange chromatography combined with bottom-up and top-down proteomics revealed the precise hierarchy of phosphorylation in this organism, where the first phosphorylated residues corresponded to the CDK consensus motifs. Finally, relative quantification of the phosphorylated species by stable-isotope labeling, Immobilized Metal Affinity Chromatography (IMAC), and mass spectrometry showed that phosphorylated H1 was more abundant in growing Tetrahymena cells than in starved cells, and also confirmed the hierarchy of phosphorylation. In yeast, another unicellular eukaryote, the protein Hho I is generally considered the equivalent to metazoan histone H1. In Hho I, only three phosphorylation sites have been identified by mass spectrometry at S141, S172, and S173 (Figure 1, Table S1). The first residue, S141, is located between the two globular domains, while S172 and S173 are located in the second globular domain, WHD2 [48].
Drosophila melanogaster is a model organism widely used to study differentiation, control of gene expression, and several diseases [49]. In Drosophila, there is only one somatic linker histone (dH1), and one germ-line specific H1 (dBigH1), which is present during early embryogenesis until the zygote genome is activated [49]. The first proteomic analysis of PTMs in dH1 found modifications in the first ten amino acids during embryonic development, consisting of N-terminal acetylation, mono- and diphosphorylation (Figure 1, Table S1) [50]. Analysis of the phosphorylated positions showed that the main phosphorylation site was S10 and that the amount of this modification decreased as the embryos matured. Another four residues, S1, S3, T7, and S8 were also found phosphorylated, albeit in lower proportions than S10.
Analysis of PTMs from cultured Drosophila S2 cells showed the presence of additional modifications including two new phosphorylated positions T19 and S67 as well as eight methylation sites, three acetylation sites, and four ubiquitylation sites (Table S1) . In some cases, more than one PTM was mapped to the same lysine residue. In this study, bottom-up proteomics was complemented with the analysis of the N-terminal domain and of the intact protein in order to determine which PTMs coexisted in the same dH1 molecule. Top-down experiments showed the existence of multiple proteoforms for dH1, containing different arrays of the PTMs identified in the bottom-up approach. Middle-down analysis of the N-terminal domain identified S8 and S10 as the positions modified in the dephosphorylated species, in agreement with the previous study . Other species were detected with combinations of Nα-terminal acetylation, mono- and dephosphorylation, and dimethylation. Tri- tetra- and pentaphosphorylated species were also detected in low proportions.
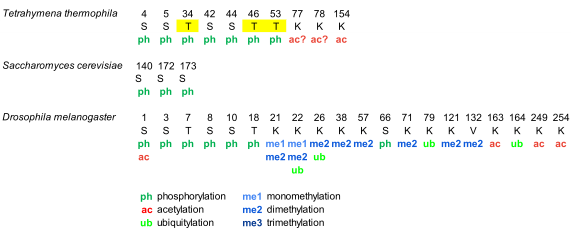
Figure 1. Modified positions in histone H1 of Tetrahymena thermophila, Saccharomyces cerevisiae, and Drosophila melanogaster. The positions refer to the mature protein, which lacks the initial methionine. Highlighted in yellow, phosphorylation in cyclin-dependent-kinase (CDK) consensus motifs. Question marks are included in post-translational modifications (PTMs) of ambiguous assignment.
2.2 PTMs identified in chicken erythrocyte linker histones.
Chicken erythrocytes are a model system to study the chromatin structure [51][52][53]. In chicken erythrocytes, there are six H1 subtypes, H1.01, H1.02, H1.03, H1.10, H1.1L and H1.1R, which amount to 40% of the linker histones. H1 subtypes have more than 85% of sequence identity and lack clear orthologous with mammalian subtypes [54]. The high sequence identity can impair the assignment of PTMs in chicken H1 subtypes because sometimes the modified peptides are shared between several subtypes. Histone H5, which is considered orthologous to mammalian H1.0, represents the remaining 60% of the linker histones .
Analysis of PTMs by bottom-up proteomics identified five modification types in chicken erythrocyte linker histones: acetylation, phosphorylation, methylation, formylation, and deamidation (Figure 2, Figure S1, and Table S2) [55][56]. All H1 subtypes were N-terminally acetylated. Two additional acetylated residues were found in the NTD of H1.02 and H1.1R (K17, for both subtypes). The NTD was also modified by phosphorylation in the first and/or the third residue, depending on the subtype. Two acetylations and one phosphorylation mapped to the GD, K34ac, K90ac, and S39p (referred to H1.01 sequence) were in peptides common to all H1 subtypes. The two of the acetylated sites of the GD were also found formylated. This modification was also detected in additional sites of the GD. The mass-shift caused by formylation is quasi-isobaric with that of dimethylation, thus, in some cases, the type of modification was not determined [56]. Additionally, one of the asparagine residues of the GD was found deamidated. Up to four acetylated sites were found in the CTD, depending on the subtype. Like in the GD, all the acetylated sites in this domain were found in peptides common to more than one subtype. Two monomethylated peptides belonging to the CTD were detected. One of these peptides is shared by two subtypes and was also acetylated. Finally, only one of the three CDK-consensus motifs in the CTD of H1 subtypes (S155, referred to H1.01) or in the CTD of H5 (S129) was phosphorylated. This result was expected as phosphorylation in H1 decreases during erythrocyte terminal differentiation [56]. In H5, most of the PTMs found were in the NTD. They consisted of five phosphorylations (T1, S3, S7, S22, and S24), three acetylations (T1, K12, and K14), and one monomethylation (K12). Furthermore, the CTD of H5 was phosphorylated in S129 and acetylated in K150. PTMs were differentially distributed among soluble and insoluble chromatin fractions, as shown by relative quantification .
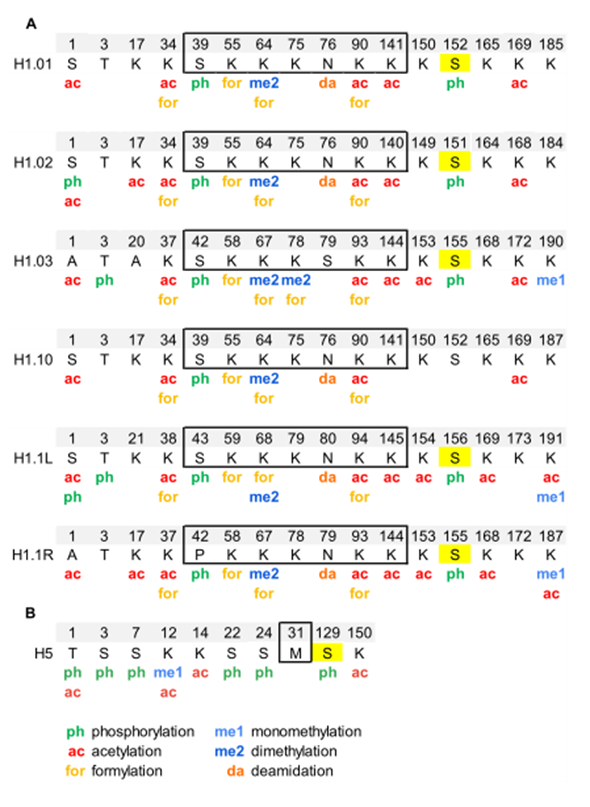
Figure 2. Modified positions in linker histones of chicken erythrocytes. (A) PTMs identified in H1 subtypes are shown in the sequence alignment. (B) PTMs identified in H5. The residues of the globular domain are shown inside the box. In yellow, phosphorylated residues located at CDK-consensus motifs. The positions refer to the mature protein, which lacks the initial methionine. The complete sequences and the original sequence alignment are shown in Figure S1.
2.3 PTMs identified in histone H1 subtypes in mammalian species.
Identification of PTMs in histone H1 in mammals has been carried out mostly in humans and mice, although some PTMs were characterized in rat testis (Figure 3, Figure S2, Table S3). The most extensive bottom-up study identified multiple PTM types in human cell lines and several mouse tissues . Other studies have targeted specific PTMs including phosphorylation , methylation , formylation , 2-hydroxyisobutyrylation, crotonylation or specific cell lines , tissues , processes [57], and subtypes [58].

Figure 3. Modified positions in mammalian H1 subtypes. The PTMs are shown based on the sequence alignment of the human, mouse, and rat sequence for each subtype. The residues of the globular domain are shown in the box. In yellow, phosphorylated residues located at CDK-consensus motifs. In blue, PTM-hotspots in the globular domain. The positions refer to the mature protein, which lacks the initial methionine. The complete sequences and the original alignments are shown in Figure S2.
From the accumulated data, several conclusions about the abundance, distribution of PTM types, and modification types can be drawn. All somatic subtypes are post-translationally modified. However, the number of modified positions appears to be determined by the abundance of each subtype. Therefore, many PTMs have been identified in the most abundant subtypes H1.2 and H1.4, while very little information is available for low abundance subtypes such as H1.0 and H1x. H1.1 has very restricted expression, but PTM mapping has been performed in the testis and in mouse embryonic stem cells (mESCs), where this subtype is present in significant proportions [59]. In some cases, PTMs in subtypes with high sequence identity, especially in the GD, like H1.2–H1.5 and to a lesser extent H1.1, might be overestimated. However, modifications in the terminal domains are often subtype-specific, as the sequence identity between subtypes is much lower in the terminal domains.
Despite the fact that new PTM types like formylation, crotonylation. or citrullination have been described, the most widespread modifications in histone H1 are phosphorylation, methylation, and acetylation. H1 subtypes contain between 27–44 residues that can be phosphorylated, of which up to 40% of them have been found to be phosphorylated (Table S4). Phosphorylated positions have been detected in all structural domains, but most of them are in the terminal domains. The NTD of subtypes H1.1–H1.5 contains two phosphorylation hotspots, the SET motif, and a CDK consensus motif or another residue phosphorylated during the cell cycle. The rest of the CDK consensus motifs (three or four, depending on the subtype) are in the CTD. Phosphorylation is highly abundant in H1, as up to 75% of the proteins are phosphorylated in mitosis [60].
All H1 subtypes are rich in basic amino acids, mostly lysine. Considering that this residue is capable of acquiring different PTMs, H1 subtypes are heavily modified. Acetylation and methylation sites are quite abundant in H1. In human cell lines, acetylation sites were more abundant than methylation, while the opposite was true for mouse and human tissues . A different study also showed that the H1 acetylation level in mESCs had increased compared to that of differentiated cells . There are also differences regarding the localization of methylation and acetylation. Lysine residues in the NTD are often methylated, whereas acetylation is predominant in the GD .
Distinct residues of mammalian H1 subtypes have been found modified by formylation, crotonylation, ubiquitylation, citrullination, 2-hydroxyisobutyrylation, and parylation (Table S3). It can be observed in Figure 3 that most of the formylated, crotonylated, and ubiquitylated sites are located in the GD in lysine residues where other PTMs have been mapped. Thus, most lysine residues of the GD including those directly involved in DNA binding can be considered PTM-hotspots as they appear to be targeted by many different modifications, which may modulate the binding properties of this domain in response to different situations and stimuli (Figure 3).
In mammals, there are four H1 germline-specific subtypes. Several studies have characterized the H1 complement in testis, thus allowing the exploration of H1 PTMs in the male germline-specific subtypes [61][62] (Figure 3, Table S3). Extensive characterization of H1 PTMs has been carried out in rat testis [61]. Perchloric acid extracts were analyzed by mass spectrometry using different separation methods, capillary electrophoresis (CESI-MS), and nano-HPLC-liquid chromatography (LC-ESI-MS/MS). This procedure allowed the identification of modifications in somatic H1 subtypes H1.0–H1.5 and one of the male germline-specific subtypes, H1t. Multiple PTMs were detected in subtypes H1.1, H1.3, H1.4, and H1t, while few modified sites were found in H1.0, H1.2, and H1.5 (Table S3). Most of the sites were identified by both CE and LC, but a few were detected by only one method [34]. The most abundant PTM was phosphorylation, but a few acetylated residues were also found. Acetylation was found at the N-terminus of all the detected subtypes, and also at other positions in subtypes H1.1, H1.3, and H1t. Multiple phosphorylated sites were detected in all subtypes including most of the CDK motifs present in subtypes H1.1, H1.3, H1.4, and H1t. In agreement with mouse and human data, most of H1t PTMs were located in the CTD, but no methylated residues were detected in this species. More PTMs were detected during mouse spermatogenesis than in mature human sperm (Table S3). In H1t, as in somatic subtypes, the predominant modification types included phosphorylation, methylation, and acetylation. In contrast, in H1t, the PTMs were mainly located in the CTD, whereas in somatic subtypes, PTMs have been mapped in the three structural domains of H1 (Figure 3).
Endogenous HILS1, another male germline-specific subtype, was separated from the rest of the H1 subtypes by reversed-phase high-performance liquid chromatography (RP-HPLC), allowing the identification of 15 PTMs including acetylation and phosphorylation. In particular, phosphorylation appeared to be abundant in this protein, as over 40% of the S/T/Y residues were found phosphorylated, with most of the phosphorylation sites located in the GD (Figure 3, Figure S2, Tables S3 and S4) . Interestingly, tyrosine phosphorylation was detected for the first time in linker histones at Y78 of this subtype, a residue located in the globular domain. HILS1Y78p appears in early elongating spermatids, where it co-localized with Transition Protein 2 (TP2), and disappears from the head region of condensing spermatids, remaining only in the tail region . While multiple PTMs were detected in human, mouse, and rat H1t as well as in rat HILS1, no post-translational modifications have been detected in the third male-germline specific subtype, H1T2.
PTMs have been detected in nine out of eleven mammalian H1 subtypes, depending on the species. At least 13 modification types have been described in H1, with phosphorylation, methylation, and acetylation the most abundant. Almost 400 positions, in the consensus sequence for each subtype, have been found modified. This number is a rough estimation of the extent of modification of H1 subtypes because some residues can have alternative PTMs, and also because promiscuous peptides are mapped to more than one subtype. However, the number of PTMs whose function has been described is quite small in comparison.
3. Summary
H1 PTMs have been characterized from lower eukaryotes to humans. The most abundant modification is phosphorylation, followed by methylation and acetylation. PTMs in H1 are variable, depending on the cell type and cell-cycle phase. Post-translational modifications in H1 may favor H1 binding or dissociation from chromatin, therefore altering chromatin compaction, which is a key factor in most nuclear processes. In organisms with more that one H1 subtype, the presence of subtype-specific PTMs can be associated with subtype functional differentiation.
References
- Bradbury, E.M.; Cary, P.D.; Chapman, G.E.; Crane-Robinson, C.; Danby, S.E.; Rattle, H.W.; Boublik, M.; Palau, J.; Aviles, F.J. Studies on the role and mode of operation of the very-lysine-rich histone H1 (F1) in eukaryote chromatin. The conformation of histone H1. Eur. J. Biochem. 1975, 52, 605–613.
- Lu, X.; Hansen, J.C. Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J. Biol. Chem. 2004, 279, 8701–8707.
- Th’ng, J.P.; Sung, R.; Ye, M.; Hendzel, M.J. H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J. Biol Chem. 2005, 280, 27809–27814.
- Roque, A.; Orrego, M.; Ponte, I.; Suau, P. The preferential binding of histone H1 to DNA scaffold-associated regions is determined by its C-terminal domain. Nucleic Acids Res. 2004, 32, 6111–6119.
- Widlak, P.; Kalinowska, M.; Parseghian, M.H.; Lu, X.; Hansen, J.C.; Garrard, W.T. The histone H1 C-terminal domain binds to the apoptotic nuclease, DNA fragmentation factor (DFF40/CAD) and stimulates DNA cleavage. Biochemistry 2005, 44, 7871–7878.
- Roque, A.; Sortino, R.; Ventura, S.; Ponte, I.; Suau, P. Histone H1 Favors Folding and Parallel Fibrillar Aggregation of the 1–42 Amyloid-β Peptide. Langmuir 2015, 31, 6782–6790.
- Kasinsky, H.E.; Lewis, J.D.; Dacks, J.B.; Ausió, J. Origin of H1 linker histones. FASEB J. 2001, 15, 34–42.
- Ponte, I.; Romero, D.; Yero, D.; Suau, P.; Roque, A. Complex evolutionary history of the mammalian histone H1.1-H1.5 gene family. Mol. Biol. Evol. 2017, 34, 545–558.
- Patterton, H.G.; Landel, C.C.; Landsman, D.; Peterson, C.L.; Simpson, R.T. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 7268–7276.
- Panday, A.; Grove, A. The high mobility group protein HMO1 functions as a linker histone in yeast. Epigenetics Chromatin 2016, 9, 13, doi:10.1186/s13072-016-0062-8. eCollection 2016.
- Talbert, P.B.; Ahmad, K.; Almouzni, G.; Ausió, J.; Berger, F.; Bhalla, P.L.; Bonner, W.M.; Cande, W.Z.; Chadwick, B.P.; Chan, S.W.L.; et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 2012, 5, 7, doi:10.1186/1756-8935-5-7.
- Millán-Ariño, L.; Izquierdo-Bouldstridge, A.; Jordan, A. Specificities and genomic distribution of somatic mammalian histone H1 subtypes. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 510–519.
- Ponte, I.; Vidal-Taboada, J.M.; Suau, P. Evolution of the vertebrate H1 histone class: Evidence for the functional differentiation of the subtypes. Mol. Biol. Evol. 1998, 15, 702–708.
- Orrego, M.; Ponte, I.; Roque, A.; Buschati, N.; Mora, X.; Suau, P. Differential affinity of mammalian histone H1 somatic subtypes for DNA and chromatin. BMC Biol. 2007, 5, 22, doi:10.1186/1741-7007-5-22.
- Clausell, J.; Happel, N.; Hale, T.K.; Doenecke, D.; Beato, M. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS ONE 2009, 4, e0007243.
- Raghuram, N.; Carrero, G.; Th’ng, J.; Hendzel, M.J. Molecular dynamics of histone H1. Biochem. Cell Biol. 2009, 87, 189–206.
- Li, J.Y.; Patterson, M.; Mikkola, H.K.A.; Lowry, W.E.; Kurdistani, S.K. Dynamic Distribution of Linker Histone H1.5 in Cellular Differentiation. PLoS Genet. 2012, 80, e1002879.
- Izzo, A.; Kamieniarz-Gdula, K.; Ramírez, F.; Noureen, N.; Kind, J.; Manke, T.; vanSteensel, B.; Schneider, R. The Genomic Landscape of the Somatic Linker Histone Subtypes H1.1 to H1.5 in Human Cells. Cell Rep. 2013, 3, 2142–2154.
- Millán-Ariño, L.; Islam, A.B.M.M.K.; Izquierdo-Bouldstridge, A.; Mayor, R.; Terme, J.-M.; Luque, N.; Sancho, M.; López-Bigas, N.; Jordan, A. Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 2014, 42, 4474–4493.
- Mayor, R.; Izquierdo-Bouldstridge, A.; Millán-Ariño, L.; Bustillos, A.; Sampaio, C.; Luque, N.; Jordan, A. Genome distribution of replication-independent histone H1 variants shows H1.0 associated with nucleolar domains and H1X associated with RNA polymerase II-enriched regions. J. Biol. Chem. 2015, 290, 7474–7491.
- Bradbury, E.M.; Inglis, R.J.; Matthews, H.R.; Sarner, N. Phosphorylation of Very-Lysine-Rich Histone in Physarum polycephalum Correlation with Chromosome Condensation. Eur. J. Biochem. 1973, 33, 131–139.
- Gorovsky, M.A.; Keevert, J.B.; Pleger, G.L. Histone F1 of Tetrahymena macronuclei: Unique electrophoretic properties and phosphorylation of F1 in an amitotic nucleus. J. Cell Biol. 1974, 1, 134–145.
- Allis, C.D.; Gorovsky, M.A. Histone Phosphorylation in Macro and Micronuclei of Tetrahymena thermophila. Biochemistry 1981, 20, 3828–3833.
- Blumenfeld, M. Phosphorylated H1 histone in Drosophila melanogaster. Biochem. Genet. 1979, 17, 163–166.
- Sung, M.T.; Wagner, T.E.; Hartford, J.B.; Serra, M.; Vandegrift, V.; Sung, M.T. Phosphorylation and Dephosphorylation of Histone V (H5): Controlled Condensation of Avian Erythrocyte Chromatin. Appendix: Phosphorylation and Dephosphorylation of Histone H5. II. Circular Dichroic Studies. Biochemistry 1977, 16, 286–290.
- Balhorn, R.; Chalkley, R.; Granner, D. Lysine-Rich Histone Phosphorylation. A Positive Correlation with Cell Replication. Biochemistry 1972, 11, 1094–1098.
- Gurley, L.R.; Walters, R.A.; Tobey, R.A. Sequential phosphorylation of histone subfractions in the Chinese hamster cell cycle. J. Biol. Chem. 1975, 250, 3936–3944.
- Moradian, A.; Kalli, A.; Sweredoski, M.J.; Hess, S. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 2014, 14, 489–497.
- Wiśniewski, J.R.; Zougman, A.; Krüger, S.; Mann, M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell. Proteom. 2007, 6, 72–87.
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Nε-Formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008, 36, 570–577.
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938.
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028.
- Sarg, B.; Lopez, R.; Lindner, H.; Ponte, I.; Suau, P.; Roque, A. Sequence conservation of linker histones between chicken and mammalian species. Data Brief. 2014, 1, 60–64.
- Sarg, B.; Faserl, K.; Kremser, L.; Halfinger, B.; Sebastiano, R.; Lindner, H.H. Comparing and combining capillary electrophoresis electrospray ionization mass spectrometry and nano-liquid chromatography electrospray ionization mass spectrometry for the characterization of post-translationally modified histones. Mol. Cell. Proteom. 2013, 12, 2640–2656.
- Starkova, T.Y.; Artamonova, T.O.; Ermakova, V.V.; Chikhirzhina, E.V.; Khodorkovskii, M.A.; Tomilin, A.N. The Profile of Post-translational Modifications of Histone H1 in Chromatin of Mouse Embryonic Stem Cells. Acta Naturae 2019, 11, 82–91.
- Starkova, T.Y.; Polyanichko, A.M.; Artamonova, T.O.; Khodorkovskii, M.A.; Kostyleva, E.I.; Chikhirzhina, E.V.; Tomilin, A.N. Post-translational modifications of linker histone H1 variants in mammals. Phys. Biol. 2017, 14, doi:10.1088/1478-3975/aa551a
- Perri, A.M.; Agosti, V.; Olivo, E.; Concolino, A.; De Angelis, M.T.; Tammè, L.; Fiumara, C.V.; Cuda, G.; Scumaci, D. Histone proteomics reveals novel post-translational modifications in breast cancer. Aging 2019, 11, 11722–11755.
- Garcia, B.A.; Joshi, S.; Thomas, C.E.; Chitta, R.K.; Diaz, R.L.; Busby, S.A.; Andrews, P.C.; Ogorzalek Loo, R.R.; Shabanowitz, J.; Kelleher, N.L.; et al. Comprehensive phosphoprotein analysis of linker histone H1 from tetrahymena thermophila. Mol. Cell. Proteom. 2006, 5, 1593–1609.
- Bonet-Costa, C.; Vilaseca, M.; Diema, C.; Vujatovic, O.; Vaquero, A.; Omeñaca, N.; Castejón, L.; Bernués, J.; Giralt, E.; Azorín, F. Combined bottom-up and top-down mass spectrometry analyses of the pattern of post-translational modifications of Drosophila melanogaster linker histone H1. J. Proteom. 2012, 75, 4124–4138.
- Zheng, Y.; John, S.; Pesavento, J.J.; Schultz-Norton, J.R.; Schiltz, R.L.; Baek, S.; Nardulli, A.M.; Hager, G.L.; Kelleher, N.L.; Mizzen, C.A. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell Biol. 2010, 189, 407–415.
- Chen, Y.; Hoover, M.E.; Dang, X.; Shomo, A.A.; Guan, X.; Marshall, A.G.; Freitas, M.A.; Young, N.L. Quantitative mass spectrometry reveals that intact histone H1 phosphorylations are variant specific and exhibit single molecule hierarchical dependence. Mol. Cell. Proteom. 2016, 15, 818–833.
- Garcia, B.A.; Busby, S.A.; Barber, C.M.; Shabanowitz, J.; Allis, C.D.; Hunt, D.F. Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry. J. Proteome Res. 2004, 3, 1219–1227.
- Lu, A.; Zougman, A.; Pudełko, M.; Bȩbenek, M.; Ziółkowski, P.; Mann, M.; Wiśniewski, J.R. Mapping of lysine monomethylation of linker histones in human breast and its cancer. J. Proteome Res. 2009, 8, 4207–4215.
- Dai, L.; Peng, C.; Montellier, E.; Lu, Z.; Chen, Y.; Ishii, H.; Debernardi, A.; Buchou, T.; Rousseaux, S.; Jin, F.; et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014, 10, 365–370.
- Christophorou, M.A.; Castelo-Branco, G.; Halley-Stott, R.P.; Oliveira, C.S.; Loos, R.; Radzisheuskaya, A.; Mowen, K.A.; Bertone, P.; Silva, J.C.R.; Zernicka-Goetz, M.; et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 2014, 507, 104–108.
- Roth, S.Y.; Schulman, I.G.; Richman, R.; Cook, R.G.; Allis, C.D. Characterization of phosphorylation sites in histone H1 in the amitotic macronucleus of Tetrahymena during different physiological states. J. Cell Biol. 1988, 107, 2473–2482.
- Mizzen, C.A.; Dou, Y.; Liu, Y.; Cook, R.G.; Gorovsky, M.A.; Allis, C.D. Identification and mutation of phosphorylation sites in a linker histone. Phosphorylation of macronuclear H1 is not essential for viability in Tetrahymena. J. Biol. Chem. 1999, 274, 14533–14536.
- Mukherjee, K.; English, N.; Meers, C.; Kim, H.; Jonke, A.; Storici, F.; Torres, M. Systematic analysis of linker histone PTM hotspots reveals phosphorylation sites that modulate homologous recombination and DSB repair. DNA Repair 2020, 86, 102763, doi:10.1016/j.dnarep.2019.102763.
- Bayona-Feliu, A.; Casas-Lamesa, A.; Carbonell, A.; Climent-Cantó, P.; Tatarski, M.; Pérez-Montero, S.; Azorín, F.; Bernués, J. Histone H1: Lessons from Drosophila. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 526–532.
- Villar-Garea, A.; Imhof, A. Fine mapping of posttranslational modifications of the linker histone H1 from Drosophila melanogaster. PLoS ONE 2008, 3, e1553.
- Thomas, J.O. The higher order structure of chromatin and histone H1. J. Cell Sci. 1984, 1, 1–20.
- Cogburn, L.A.; Porter, T.E.; Duclos, M.J.; Simon, J.; Burgess, S.C.; Zhu, J.J.; Cheng, H.H.; Dodgson, J.B.; Burnside, J. Functional genomics of the chicken-A model organism. Poult. Sci. 2007, 86, 2059–2094.
- Kowalski, A.; Pałyga, J. Chromatin compaction in terminally differentiated avian blood cells: The role of linker histone H5 and non-histone protein MENT. Chromosom. Res. 2011, 19, 579–590.
- Koutzamani, E.; Loborg, H.; Sarg, B.; Lindner, H.H.; Rundquist, I. Linker histone subtype composition and affinity for chromatin in situ in nucleated mature erythrocytes. J. Biol. Chem. 2002, 277, 44688–44694.
- Sarg, B.; Lopez, R.; Lindner, H.; Ponte, I.; Suau, P.; Roque, A. Identification of novel post-translational modifications in linker histones from chicken erythrocytes. J. Proteom. 2015, 113, 162–177.
- Snijders, A.P.L.; Pongdam, S.; Lambert, S.J.; Wood, C.M.; Baldwin, J.P.; Dickman, M.J. Characterization of post-translational modifications of the linker histones HI and H5 from chicken erythrocytes using mass spectrometry. J. Proteome Res. 2008, 7, 4326–4335.
- Luense, L.J.; Wang, X.; Schon, S.B.; Weller, A.H.; Lin Shiao, E.; Bryant, J.M.; Bartolomei, M.S.; Coutifaris, C.; Garcia, B.A.; Berger, S.L. Comprehensive analysis of histone post-translational modifications in mouse and human male germ cells. Epigenetics Chromatin 2016, 9, 24, doi:10.1186/s13072-016-0072-6. eCollection 2016.
- Mishra, L.N.; Gupta, N.; Rao, S.M.R. Mapping of post-translational modifications of spermatid-specific linker histone H1-like protein, HILS1. J. Proteom. 2015, 128, 218–230.
- Pan, C.; Fan, Y. Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 496–509.
- Talasz, H.; Helliger, W.; Puschendorf, B.; Lindner, H. In vivo phosphorylation of histone H1 variants during the cell cycle. Biochemistry 1996, 35, 1761–1767.
- Rose, K.L.; Li, A.; Zalenskaya, I.; Zhang, Y.; Unni, E.; Hodgson, K.C.; Yu, Y.; Shabanowitz, J.; Meistrich, M.L.; Hunt, D.F.; et al. C-terminal phosphorylation of murine testis-specific histone H1t in elongating spermatids. J. Proteome Res. 2008, 7, 4070–4078.
- Sarg, B.; Chwatal, S.; Talasz, H.; Lindner, H.H., Testis-specific linker histone H1t is multiply phosphorylated during spermatogenesis. Identification of phosphorylation sites. J. Biol Chem. 2009, 284, 3610–3618.


