 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura-Ancuta Pop | + 1861 word(s) | 1861 | 2020-08-17 08:19:30 | | | |
| 2 | Conner Chen | Meta information modification | 1861 | 2020-08-24 11:04:10 | | |
Video Upload Options
Genome-wide association studies (GWAS) are studies assessing and analyzing either differences or variations in DNA sequences across the human genome to detect genetic risk factors of diseases prevalent within a target population under study.
1. Introduction
It is known that GWAS assess and analyze differences or variations in DNA sequences across the human genome to detect genetic risk factors of diseases prevalent in a population under investigation. The ultimate goal of GWAS is to predict either disease risk or disease progression by utilizing genetic risk factors to define the biological basis of disease susceptibility. This also enables the development of innovative and preventative therapeutic strategies [1]. There are two fundamental concepts underlying GWAS, including linkage disequilibrium (LD) and a common disease–common variant (CD–CV) hypothesis. A basic technical workflow for a typical GWAS is presented in Figure 1.
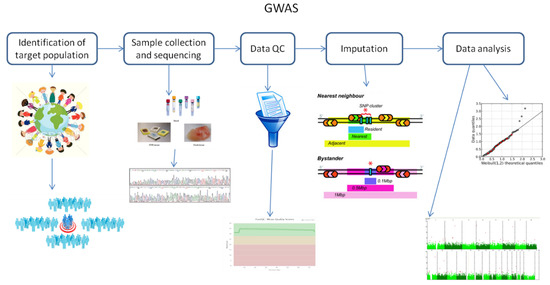
Figure 1. A technical flow chart for genome-wide association studies (GWAS). Abbreviation: QC, quality control.
Therefore, GWAS serve as a tool to identify associations between genetic regions and specific traits of interest. A basic GWAS will evaluate the genetic profiles of hundreds of patients of a well-defined phenotype to those of hundreds of control subjects.
2. Fundamental Principles of Genome-Wide Association Studies
As mentioned above, there are two fundamental concepts underlying GWAS. These are linkage disequilibrium (LD) and the common disease–common variant (CD–CV) hypothesis.
LD is defined as a non-random association of two alleles at two or more loci [2]. In turn, this provides insights into past genetic conditions and constraints, allowing for the determination of whether selection was either natural; epigenetic [3]; or owing to other mechanisms that do not occur in isolation, such as genetic drift or gene flow. If detected throughout the genome when investigating populations, LD mirrors population history, breeding system, and geographic subdivision patterns. When investigating genomic regions, LD reveals history of natural selection, gene conversion, mutation, and other forces that either contribute to or cause gene-frequency evolution. Therefore, detecting LD does not guarantee either linkage or lack of equilibrium. Ultimately, it is the local recombination rate that determines how the aforementioned factors affect LD within a certain genomic region or between paired loci [2]. As to be expected, this is related to the concept of chromosomal linkage, which infers that two markers found on a chromosome will remain physically linked on that chromosome throughout consecutive familial generations. However, recombination events will separate chromosomal segments within a family from one generation to another, and this effect is continuously amplified through several subsequent generations. Inevitably, recombination events will break apart segments of chromosomes carrying linked alleles until all alleles within a population are in linkage equilibrium. Simply stated, linkage disequilibrium involves coupling markers at the population level [1]. Furthermore, the rate of LD decay is dependent on the following factors: population size, number of founding chromosomes within a population, and number of generations for which the population has existed. Therefore, it comes as no surprise that there are different levels and patterns of LD when comparing different human subpopulations. For example, the most ancestral human population is that of an African-descent population, which, owing to the accumulation of more recombination events, has smaller regions of LD. Meanwhile, on average, European-descent and Asian-descent populations have larger regions of LD than African-descent populations. This is attributed to the fact that European- and Asian-descendant populations have been generated by founding events, whereby they have split from the African population, thus inherently changing the number of founding chromosomes, population size, and generational age of the population [1].
In general, closely linked polymorphic SNPs have strong LD between them. The International HapMap consortium has demonstrated that the human genome contains haplotype blocks, within which either most or all are high LD SNPs. Thus, there is a fine-scale pattern of LD present in human populations. Subsequently, it has been assumed that high LD levels detected among SNPs are for those alleles that exhibit increased risks of complex inherited diseases [2]. Interestingly, this has been in fact observed for those SNPs significantly associated with breast cancer when GWAS have been conducted and large numbers of SNPs have been surveyed [4]. However, it is important to take into consideration that LD in GWAS can be generated by either undetected or unknown population stratification. Moreover, GWAS have been successful at uncovering associated SNPs, despite the low overall breast cancer risk within a population, and even in identifying new causative alleles [2]. In fact, five new variants have been found to be associated with familial breast cancer, but only 3.6% of familial breast cancer can be attributed to these alleles [4]. However, it is important to point out that it is the relatively high frequency of these causative alleles that allows for their detection by GWAS.
The CD–CV hypothesis [5] has been developed based on the following two principles: common diseases differ from rare disorders in terms of their underlying genetic architecture, and the discovery of several susceptibility variants for a common disease is of high minor allele frequency [1]. In other words, this hypothesis proposes that common diseases are influenced by genetic variations common within a population [5]. Firstly, this suggests that there has to be a high correlation between allele frequency and population occurrence. Secondly, if common genetic variants influence disease, then the effect size or penetrance for any one variant must be small relative to those exhibited by rare diseases [1]. This further implies that, if the same SNP causes a small change in gene expression that alters disease risk by a small proportion, this creates a scenario wherein the frequency of disease incidence and the causal allele are only lowly-correlated. Thus, common variants cannot yield high effects. Thirdly, disease susceptibility is influenced by multiple common alleles based on the condition that common alleles have small genetic effects, and that common diseases exhibit heritability. Additionally, if an allele of a single SNP slightly increases disease risk, this implies that such an SNP accounts for a small amount of variance of the total variation caused by genetic factors. Consequently, multiple genetic factors synergistically account for the total genetic risk of a common genetic variation [1]. However, it is important not to jump to the conclusion that the entire genetic component of any disease is attributable only to common alleles.
3. Challenges of Genome-Wide Association Studies
While discussing genetic heterogeneity and the potential role of rare genetic variants in complex human diseases, McClellan and King [6] have pointed out some important and interesting criticisms of GWAS. Despite the fact that some of these criticisms have already been addressed, it is important to go through them to better understand these issues, and to improve the outcomes of GWAS.
One of these noted issues pertains to the fact that some of the genetic variants lack biological functions, and thereby their relative importance is highly diminished. In fact, it has been observed that GWAS are populated by risk variants of no known functions. Thus, the utility and reliability of GWAS have been questioned as most detected SNPs in GWAS are from intergenic regions [6]. Furthermore, GWAS identify approximate locations of loci associated with disease variants rather than attempt to specifically identify functional SNPs. This is attributed to widespread LD between segregating sites within a given human population. In addition, most SNPs in SNP arrays have unknown biological functions, as most SNPs in HapMaps are located in noncoding regions, and SNP arrays usually do not select for SNPs of known functions. Moreover, it is also important to emphasize that GWAS variants are not functional variants that confer risk, also referred to as “risk variants” in the published literature. Thus, 100% of a subpopulation carrying a risk allele does not truly suggest that all subjects of such a population are predisposed to risk. This simply indicates that LD patterns at a target locus are different than those of another subpopulation. Although the majority of thousands of “risk variants” that have been identified from GWAS have no apparent known biological functions, these are explained by using deduction and rationale, as outlined by the CD–CV hypothesis. This suggests that most genotyping platforms select for common variants. Moreover, as evolution has ensured that the most common variants are neutral, it should come as no surprise that most GWAS findings are neutral, originating from factors other than associations with disease risk. On the basis of evolutionary genetics, most alleles are in fact recent, and they are rare [7][8]. It is unclear what is exactly required for a common allele to remain in a population, as mechanisms of evolution can both facilitate and hinder heritability, particularly as they do not occur in isolation. For example, an allele can significantly increase in frequency without any need for selection when either a population bottleneck occurs (genetic drift) or when a subpopulation migrates and integrates with another (gene flow).
In another claim, it has been reported that it is population stratification that results in GWAS hits [6]. Although population stratification or substructure inflates test statistics, this can be readily identified, and adjusted for accordingly. In general, populations differ among each other over many loci and not only for one or two SNPs, which is precisely how whole-genome data are used to identify stratification. This is exemplified by the particularly fine-scale sub-populations in Europe that can be readily separated utilizing whole-genome data. Most importantly, as population stratification is one of the fundamental assumptions taken into consideration by the CD–CV hypothesis, the GWAS community has established methods to deal with population stratification that are fairly effective for common variants. For example, EigenStrat is a multi-dimensional scaling approach for addressing stratification, and is commonly used as a standard practice in the case-control GWAS dataset. Additionally, family-based study designs in GWAS have an advantage in protecting against stratification. Lastly, frequency estimates are dependent on sample size, thus conferring additional variations to such results [7][8].
As with all studies, sample size significantly impacts interpretations of data. Single GWAS analyses are relatively underpowered owing to the fact that they have a limited number of samples, which drastically increases the probability of false-positive findings. Given this, implementing meta-analysis of several GWAS can overcome these small-sample numbers and study-specific limitations, thus providing a more robust statistical analysis and reduced false-positive results. To date, there are many published articles describing the meta-analysis of GWAS [9][10][11]. However, each meta-analysis consists of several stages comprised of analysis set-up, investigating heterogeneity, data storage, and variant selection for any subsequent analysis. There are several parameters and methods employed for meta-analyses, such as p-values, fixed effects, random effects, Bayesian statistics, and multivariate analysis [12]. Using such meta-analysis methods, a new collaboration, iGOGS, has discovered 74 new susceptibly loci for hormone-dependent cancers [13][14][15][16]. However, there are other consortia that have used this method for identifying other SNPs relevant for each type of disease, such as BCAC [17], ISC [18], and MAGIC [19].
The use of GWAS for cancer research studies has encountered several challenges, including the following: sample size; high numbers (430) of significant SNPs for cancer; association of several SNPs with multiple cancer localizations; implications of identified genes in several key signaling pathways involved in cancer; modulation of some pathways by lifestyle and environment; and, lastly, the fact that most studies are conducted using European populations, thus limiting extrapolation of these findings to other populations [20].
References
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822, doi:10.1371/journal.pcbi.1002822.
- Slatkin, M. Linkage disequilibrium—Understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485, doi:10.1038/nrg2361.
- Sachs, T. Epigenetic selection: an alternative mechanism of pattern formation. J. Theor. Biol. 1988, 134, 547–559, doi:10.1016/s0022-5193(88)80056-0.
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093, doi:10.1038/nature05887.
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. TIG 2001, 17, 502–510.
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217, doi:10.1016/j.cell.2010.03.032.
- Wang, K.; Bucan, M.; Grant, S.F.; Schellenberg, G.; Hakonarson, H. Strategies for genetic studies of complex diseases. Cell 2010, 142, 351–353; author reply 353–355, doi:10.1016/j.cell.2010.07.025.
- Klein, R.J.; Xu, X.; Mukherjee, S.; Willis, J.; Hayes, J. Successes of genome-wide association studies. Cell 2010, 142, 350–351; author reply 353–355, doi:10.1016/j.cell.2010.07.026.
- Zeggini, E.; Ioannidis, J.P. Meta-analysis in genome-wide association studies. Pharmacogenomics 2009, 10, 191–201, doi:10.2217/14622416.10.2.191.
- Panagiotou, O.A.; Willer, C.J.; Hirschhorn, J.N.; Ioannidis, J.P. The power of meta-analysis in genome-wide association studies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 441–465, doi:10.1146/annurev-genom-091212-153520.
- Dimou, N.L.; Tsirigos, K.D.; Elofsson, A.; Bagos, P.G. GWAR: Robust analysis and meta-analysis of genome-wide association studies. Bioinformatics (Oxford, England) 2017, 33, 1521–1527, doi:10.1093/bioinformatics/btx008.
- Evangelou, E.; Ioannidis, J.P. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013, 14, 379–389, doi:10.1038/nrg3472.
- Garcia-Closas, M.; Couch, F.J.; Lindstrom, S.; Michailidou, K.; Schmidt, M.K.; Brook, M.N.; Orr, N.; Rhie, S.K.; Riboli, E.; Feigelson, H.S.; et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet. 2013, 45, 392–398, 398e391-392, doi:10.1038/ng.2561.
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat. Genet. 2013, 45, 385–391, 391e1–391e2, doi:10.1038/ng.2560.
- Pharoah, P.D.; Tsai, Y.Y.; Ramus, S.J.; Phelan, C.M.; Goode, E.L.; Lawrenson, K.; Buckley, M.; Fridley, B.L.; Tyrer, J.P.; Shen, H.; et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat. Genet. 2013, 45, 362–370, 370e1–370e2, doi:10.1038/ng.2564.
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361, 361e1–361e2, doi:10.1038/ng.2563.
- Consortium, T.B.C.A. BCAC. University of Cambrige: Cambrige, UK. Available online: http://bcac.ccge.medschl.cam.ac.uk/ (accessed on 10 July 2020).
- Brigham, M.G. ISC. Mass General Brigham: Boston, MA, USA. Available online: https://www.massgeneral.org/ (accessed on 10 July 2020).
- Consortium), t.M.-A.o.G.a.I.-r.t. MAGIC. Sanger Institute: Cambrige, UK. Available online: https://www.sanger.ac.uk/ (accessed on 10 July 2020).
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704, doi:10.1038/nrc.2017.82.



