 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Suresh Ramakrishna | + 8196 word(s) | 8196 | 2020-08-07 11:22:17 | | | |
| 2 | Camila Xu | + 220 word(s) | 8416 | 2020-08-14 06:23:01 | | | | |
| 3 | Camila Xu | + 221 word(s) | 8417 | 2020-08-14 06:29:51 | | | | |
| 4 | Camila Xu | + 221 word(s) | 8417 | 2020-08-14 06:30:41 | | | | |
| 5 | Suresh Ramakrishna | + 216 word(s) | 8412 | 2020-08-14 06:31:20 | | | | |
| 6 | Camila Xu | -4787 word(s) | 3625 | 2020-10-27 07:25:31 | | | | |
| 7 | Camila Xu | -4785 word(s) | 3627 | 2020-10-27 07:26:14 | | |
Video Upload Options
Phenylalanine hydroxylase (PAH) and fumarylacetoacetate hydroxylase (FAH) are two highly regulated liver enzymes that catalyze the rate-limiting step in phenylalanine and tyrosine metabolism. Mammalian PAH (phenylalanine 4-monooxygenase, E.C. 1.14.16.1) catalyzes the stereospecific hydroxylation of L-phenylalanine into L-tyrosine using tetrahydrobiopterin (BH4), non-heme iron, and dioxygen as co-substrates in the cytosol of the liver and kidney. PAH facilitates oxidation of excess L-phenylalanine into carbon dioxide and water, and is the major enzyme degrading 75% of L-phenylalanine from the diet. PAH assembles as a homotetrameric protein, each subunit composed of N-terminal regulatory domain for allosteric activation by Phe, a central catalytic domain, and C-terminal helix responsible for tetramer formation.
1. Introduction
Fumarylacetoacetate hydroxylase (FAH) is the last enzyme in the tyrosine catabolism pathway, and it catalyzes the hydrolysis of fumarylacetoacetate into fumarate and acetoacetate as the final step in phenylalanine and tyrosine degradation. FAH is a cytosolic dimer that consists of two α–β domains; 300 residues of the C-terminal domain form the active site that binds to Ca2+ and participates in intermolecular interactions at the dimer interface; 120 residues of the N-terminal domain play the regulatory role [6,7]. The FAH dimer is solely considered to be catalytically active [7]. The human FAH gene occupies chromosome 15q23–q25, spans 30–35 kb, and contains 14 exons [8], whereas the PAH gene is located on chromosome 12q at position 23.2, spans 90 kb, and contains 13 exons [9].
In 1932, Grace Medes discovered 4-hydroxyphenylpyruvate in the urine of a 49-year-old man and described it as “tyrosinosis” [10]. In the 1960s, the condition was referred to as hereditary tyrosinemia type-I (HT1), and it was later understood to result from FAH deficiency [11,12,13,14]. Deficiency of this enzyme leads to the accumulation of upstream metabolites such as fumarylacetoacetate (FAA) and maleylacetoacetate, which are subsequently converted into succinylacetone. FAA and succinylacetone are both genotoxic and carcinogenic [15]. Similarly, in 1934, Dr. Asbjørn Følling recognized elevated levels of phenylketonuric acid in the urine of two mentally retarded siblings and named the condition “phenylpyruvic oligophrenia” or phenylketonuria (PKU) [3]. Elevated levels of blood phenylalanine and its metabolites, such as keto acid and phenylpyruvate, along with reduced blood tyrosine levels, are the characteristics of PKU and its milder variant hyperphenylalaninemia (HPA). PKU is classified as classical PKU (plasma Phe levels > 1200 μM), mild or atypical or variant PKU (600–1200 μM), and non-PKU mild HPA (120–600 μM) [16,17]. PKU is associated with mental retardation, epilepsy, brain damage, and neurological and behavioral problems due to the accumulation of phenylalanine byproducts. Tyrosine is the precursor for multiple molecules; therefore, tyrosine deficiency leads to deficiency of catecholamine neurotransmitters, melanin, and L-thyroxine [3,18].
HT1 pathogenicity is largely unknown; however, missense mutations in the FAH gene may influence catalytic activity, protein stability, and/or protein homeostasis and monomer-dimer equilibrium [7]. Despite being studied extensively since years, the pathophysiology of PKU is not fully elucidated. Mutation-driven PAH protein instability, misfolding, and aggregation are the hallmark associated with the disease resulting in subsequent protein turnover [19,20,21]. The regulation of L-Phe by PAH is a complex mechanism associated with transition between oligomeric state, changes in conformation, phosphorylation and substrate activation, and cofactor inhibition [4,5]. The newly discovered crystal structure supports the notion that PAH exists in two native states: resting state-PAH (RS-PAH) and activated-PAH (A-PAH). The RS-PAH and A-PAH was determined by X-ray crystallography and small-angle X-ray scattering respectively [4]. The RS-PAH has low affinity for Phe and helps maintain the basal level of Phe essentially available for cellular functions. Also, BH4 is complexed with RS-PAH, thus acting as a negative regulator for L-Phe activation [4,22]. BH4 serves as a pharmacological chaperone stabilizing PAH and increasing the steady state level of enzyme [20]. As the concentration of Phe increases, excess Phe acts as an activator and binds to A-PAH allosterically, shifting the equilibrium from RS-PAH to A-PAH. Binding of Phe induces large conformational change and dimerization of regulatory domain of the enzyme, thus exposing the active site for the conversion of Phe to Tyr [4,5,22]. BH4 and Phe binding drives the newly synthesized, partially folded, PAH into equilibrium of native structure [22]. PKU disease-associated alleles affect several different operations (like allosteric activation by Phe, stabilization by BH4) that join forces for efficient degradation of excess Phe. Therefore, it is important to maintain the PAH structure equilibrium which is hampered due to disease-associated mutations.
The crystal structure of the PAH tetramer providing information about PAH allostery and BH4 associated stability was recently discovered [22,23]. The allosterically activated form of PAH is majorly responsible for the conversion of phenylalanine to tyrosine; however, stability calculations are not possible for this form as its high resolution structure is not yet available. Nonetheless, certain experimental reports suggested increased aggregation, high instability, and accelerated degradation of the PAH mutant expressed in Enu1/1 and Enu1/2 heteroallelic mouse model, primary hepatocytes and COS-7 cells [24,25,26]. The mutant PAH proteins (e.g., p.V106A) expressed in Enu1/1 mouse model, are also known to be highly ubiquitinated in vitro and in vivo, targeting it for proteasome-mediated degradation and selective autophagy [24].
To combat the pathogenic accumulation of defective proteins, the cells are equipped with the protein quality control (PQC) system, mainly including molecular chaperones and the ubiquitin proteasomal system (UPS). The supplementation with cofactor BH4, also acting as a pharmacological chaperone, stabilizes the PAH tetramer structure, providing a rationale for the BH4-responsive PAH-variants [20]. When PAH variants are co-expressed with GroEL/ES bacterial chaperone in Escherichia coli, decrease in dimer portions, increase in tetramer formation, and increase in residual activity were observed. These results suggest that co-expression with GroEL/ES bacterial chaperone might affect the PAH folding in Escherichia coli [19]. These results indicate that molecular chaperones have the potential to prevent protein misfolding and help to stabilize a range of mutant proteins. The proteins that cannot be stabilized by chaperones undergo degradation to avoid its interaction with other native and non-native proteins [27]. UPS is the major cellular degradation pathway, responsible for degrading more than 80% of intracellular proteins [28]. The proteins have to be tagged with ubiquitin moiety, in order to be degraded by the UPS. The PAH and FAH protein is reported to be ubiquitinated [7,24,28,29,30] and the variants are prone to aggregation and/or degradation [22]. However, the PAH and FAH variants exert some amount of residual activity depending upon the severity of mutation. Therefore, certain PAH and FAH mutants with folding defects are still functional, but they nonetheless suffer rapid degradation [21,31,32,33]. The degradative system therefore needs a way to differentiate between lethal defects and negligible defects. In this review, we discuss different strategies for stabilizing and increasing the concentrations of those functional mutant proteins, that display instability and folding defects and which are conjugated with ubiquitin molecule for degradation. We propose recruiting members of the ubiquitin proteasomal system (UPS) and protein quality control (PQC) chaperones into therapeutic endeavors to rescue functional misfolded proteins from accelerated degradation.
2. Etiology
More than 1000 mutations result in PKU [16], and more than 100 mutations result in HT1, and most of them are missense mutations [7]. In the PAH gene, 60.5% of mutations are missense mutations, and in the FAH gene, 45% of mutations are missense mutations [8,9]. The severity of a mutation depends on its effect on the resulting enzyme’s conformation and function. In other words, the genotypic effect on the clinical phenotype is variable [3,33]. Most of the mutations causing PKU and HT1 result in PAH and FAH instability, leading to misfolding and loss of function [20,22,33,34,35].
Because both enzymes are biallelic, it is possible to have many disease-causing mutations, resulting in a compound heterozygote [36,37]. Mutations in the PAH and FAH genes are known to decrease catalytic activity and reduce the kinetic stability of the enzymes, inducing accelerated degradation [7,38].
3. Epidemiology
PKU and HT1 are autosomal recessive traits that affect 1 in 2500 to 100,000 births [9,39] and 1 in 100,000 births [40], respectively. PKU has high prevalence in the United Kingdom, Turkey, and Ireland and is rare in Thailand, whereas HT1 is present worldwide except for Central America and Oceania. The most common mutation in PKU is p.Arg408Trp, which is frequently found in Russia and East European countries such as Hungary, the Czech Republic, Slovakia, and Croatia [41,42] and Baltic countries such as Estonia, Lithuania, and Latvia [42]. Mutations such as p.Arg241Cys, p.Arg243Gln, and Ex6-96A>G are frequent in the Chinese population [43], and p.Pro281Leu is common in Iranians [44]. Other common variants include p.Arg261Gln, p.Tyr414Cys, and p.Ser349Pro [42]. More than 40 mutations have been found in the FAH gene, and some of the most common are D233V in Turks, W262X in Finns [45], p.Gly64His in Asians, p.1 Met>Val in Saudi Arabians [8], and c.974C>T in Europeans and Caucasians [46].
4. Protein Quality Control
Eukaryotic cells possess a robust complement of proteins to monitor and maintain a healthy proteome for their survival. Proteome integrity is maintained under the scrutiny of the PQC system [47]. The maturation from a nascent polypeptide to a functional protein is crucial to its function and involves a multistep process, including proper post-translational modification. The folding process for some proteins starts during their synthesis itself, which is called co-translational folding, whereas other proteins fold in the cytoplasm or the endoplasmic reticulum (ER) and mitochondria after synthesis [48]. The fundamentals of protein folding are also governed by the cellular environment and its over-crowding [49]. The hydrophobic patches in a polypeptide are buried in the native state. Exposure leads to the formation of intermediates that can interact inappropriately with other molecules. Thus, several studies suggest that protein folding is initiated by composing a folding nucleus within a primary structure around which the remaining polypeptide folds. The most important requirement for a correct folding pattern is the interaction between the hydrophobic and polar residues during nucleation, which encourages the structure to be packed correctly [50].
The protein-folding mechanism is much more complex for larger proteins than for smaller ones. Evidence from various studies indicates that, during protein folding, some proteins attain the native structure, whereas others cannot, for reasons such as a non-native interaction that leads to intermediates or a transiently folded protein state. Therefore, large proteins are assembled from diverse segments or domains that are folded simultaneously and independently, ensuring the proper folding of each segment, so that they can correctly interact with one another to form a highly stable and compact, native, three-dimensional protein structure. In other words, for large protein complexes such as proteasomes and ribosomes, the folding pathway involves a two state mechanism [51,52].
The ability of a protein to fold correctly de novo, though thermodynamically favorable, is often hampered by transcriptional or translational errors, destabilizing mutations, or stress conditions such as heat, oxygen radicals, aging, or environmental threats, giving rise to misfolded proteins and off-pathway aggregates [53,54,55]. The misfolded proteins can exhibit either loss of function, characterized by protein dysfunction and a propensity for degradation, or gain of function, characterized by protein aggregates that cause the misfolding of other proteins through inappropriate interactions [50,56]. Cells are rescued from the dangers of misfolded proteins by the PQC system, which keeps proteins under the constant surveillance of molecular chaperones and induces the rapid degradation of misfolded proteins through the UPS or autophagy-driven lysosomal proteolysis [57,58] (Figure 1). PQC relies on three parallel strategies whereby misfolded proteins are refolded, degraded, or delivered to a quality-control compartment capable of sequestering them, such as the juxta nuclear quality control, insoluble protein deposit, aggresome, or ER-associated degradation (ERAD) vesicles [48].
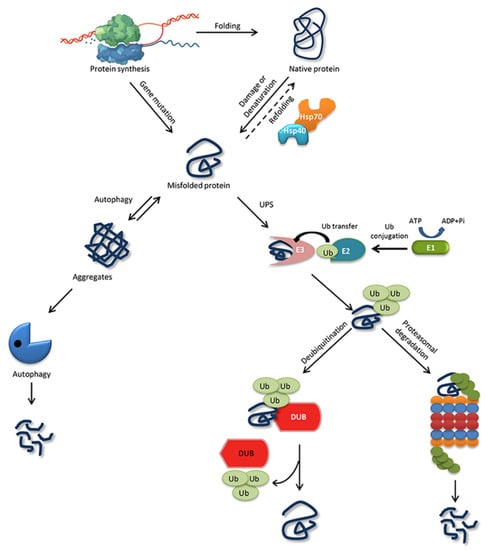
Figure 1. Protein folding, misfolding, and degradation. Protein folding starts during the ribosomal translation process and attain the native conformation to execute cellular processes. The native folded proteins are often misfolded due to mutations and other environmental factors. Molecular chaperones catalyze the folding/refolding events, disaggregation of the protein aggregates, and targeting the protein for degradation. Aggregates are typically degraded by autophagy, whereas the ubiquitin proteasome system (UPS) degrades the destabilized/misfolded proteins by covalent attachment of a ubiquitin molecule assisted by E1-E2-E3 enzymes. The ubiquitinated proteins are recognized by the 26S proteasome and are degraded. However, the ubiquitin moiety is cleaved off by Deubiquitinating enzymes (DUBs) and the protein can be rescued from the degradation cycle.
5. Deubiquitinating Enzymes Regulate Molecular Chaperones
Approximately 100 putative DUBs have been identified in humans. These large ubiquitin-cleaving proteases are classified into seven families: ubiquitin-specific proteases, ubiquitin C-terminal hydrolases, ovarian tumor proteases, Machado-Joseph disease domain proteases, Jab1/Mpn/Mov34 metalloenzymes, monocyte chemotactic protein-induced proteases, and zinc finger with UFM1-85 specific peptidase domain proteins, but most of their functions and substrates have not yet been characterized [84]. Some of the important functions of DUBs in the ubiquitin pathway include generating free ubiquitin monomers by processing inactive ubiquitin precursors, acting as an E3 ligase antagonist by cleaving the ubiquitin molecule from the substrate proteins, and maintaining a ubiquitin pool by recycling cleaved ubiquitin molecules. DUBs are known to be involved in physiological processes and thus are predicted to be involved in cancers [85], neurodegeneration [84], and infectious diseases [86]. Interestingly DUBs also regulate the members of another major degradation pathway in PQC called autophagy. Misfolded proteins are recognized by molecular chaperones in the HSP family, which coordinates with the UPS for protein refolding and the removal of misfolded proteins [87].
Ubiquitination and deubiquitination both play crucial roles in the dynamic regulation of different stages of the autophagic process. To induce proper autophagy, post-translational modification of its initiators is essential. It is a well-organized game of “on” and “off” between the E3 ligase and DUBs in controlling autophagy signals [87]. Numerous E3-chaperone complexes work in parallel to target misfolded proteins. For example, the E3 ligase carboxy-terminus of Hsc70 interacting protein (CHIP) tightly regulates the function of Hsp70/Hsp90 to orchestrate cellular protein folding and degradation. Ubiquitination of substrate proteins is antagonized by DUBs, allowing misfolded proteins to escape from degradation [88]. A growing body of evidence suggests crosstalk between the DUBs and the HSPs as well. For instance, proteasome-bound USP14 protein was found to interact with molecular chaperone Hsc70 to modulate autophagy in neuroblastoma cells. Striatal neuronal cells expressing mutant huntingtin protein had a defect in autophagosome maturation that was influenced by Hsc70 and proteasome free-USP14, indicating a link between the proteasome-independent function of USP14 and Hsc70 in mediating crosstalk among autophagy, ER stress signaling, and the proteasome [89]. Similarly, the DUB USP19 has two major isoforms. One isoform contains a transmembrane domain at its C-terminus and is associated with ERAD for an unfolded protein response; the other isoform contains an EEVD extension at the C-terminus that interacts with CHIP. The N-terminus of both isoforms interacts with the Hsp90 chaperone. The regulatory function of USP19 was recently confirmed in a study demonstrating that USP19 interacts directly with chaperone Hsp90 and upregulates the aggregation of poly-Q containing the proteins Ataxin-3 and Huntingtin, which causes spinocerebellar ataxia type-3 and Huntington’s disease, respectively [90]. Direct evidence indicates that chaperone Hsp90 enhances USP19 DUB activity by promoting its substrate recognition [91]. Hsp90 recruits misfolded proteins for refolding, and should the protein fail to refold, the co-chaperone CHIP ubiquitinates the misfolded protein for degradation with the help of Hsp90, or the misfolded protein is deubiquitinated by USP19, allowing it to avoid degradation and promoting aggregation [90]. This process is perfectly synchronized as a defense mechanism against proteins whose aggregation is cytotoxic to the cells. However, to enhance the rescue of functional mutant proteins, understanding the regulatory mechanism of DUBs and molecular chaperones is beneficial.
6. Rapid Degradation of Misfolded PAH and FAH Proteins
More than 1000 variants in the human PAH gene are recorded in the locus-specific database PAHvdb (http://www.biopku.org/home/pah.asp), and certain missense mutations in the regulatory and catalytic domains cause protein instability and folding defects of the PAH protein, resulting in its rapid degradation and loss of function [31,32,92]. Thus, PKU was generally considered to be the paradigm of misfolded metabolic diseases [93]. The destabilized mutants of PAH are precisely degraded by the cellular PQC system. Mutation-dependent destabilization and accelerated proteolytic degradation are the main pathogenic mechanisms in PKU [94]. PAH is reported to be a substrate for Ub-conjugating enzyme and is likely degraded by the UPS. Døskeland et al. demonstrated that PAH isolated from rat liver is conjugated with mono- and multi-/poly-ubiquitination at its catalytic domain [29]. More recently, in an ENU1/2 heteroallelic mouse model of HPA, mutant PAH was highly ubiquitinated, which corresponded with an increased rate of degradation [24]. The mutant proteins were degraded more rapidly than the wild type enzyme [62]. The wild type is reported to have a half-life of 2 days in rat liver and 7–8 h in hepatoma cells; in contrast, mutants are degraded rapidly, due to the destabilization of their protein structure [29]. Molecular chaperones such as DNAJC12/HSP70 play a role in processing mutant PAH for UPS-mediated degradation or ubiquitin-mediated autophagy [28,30].
Likewise, more than 100 mutations of the FAH gene cause HT1. Like PAH, most of the mutations produce FAH destabilization, causing the enzyme to be rushed to the aggregation pathway. When cells expressing the FAH protein were subjected to the proteasomal inhibitor MG132, FAH protein levels were restored. Therefore, the FAH protein undergoes proteasomal degradation [7]. FAH is also conjugated with Ub at multiple lysine residues according to the PhosphoSitePlus (www.phosphosite.org) database. However, no evidence indicates the type of ubiquitin linkage and whether it targets FAH for degradation or tags it for further cellular processes. The reduced activity and deficiency of FAH found in HT1 could result from the rapid degradation of destabilized mutant proteins [95], similar to PAH in PKU.
7. Residual Catalytic Activity of PAH and FAH Can Be Rescued by Deubiquitination or Molecular Chaperones
Certain cases of PKU result from genetic mutations that impede the normal folding of the wild type PAH protein, leading to reduced or no enzyme activity. Genotype-based prediction of metabolic phenotypes, including patients with homozygosity and those with functional hemizygosity, has been studied for several years [16]. Two alleles, both with severe mutations in the PAH gene, produce an enzyme with little or no enzyme activity, whereas the presence of two mild mutations or one severe and one mild mutation produces high residual enzyme activity, producing HPA or mild PKU (>30% activity compared with wild type PAH) [93]. Certain combinations of mutations in the genotype and their predicted residual enzyme activity have already been reported [96,97]. Some mutations characterized by high residual activity were found to be responsive to natural co-factor BH4 [98]. BH4 responsiveness has a multifactorial basis, including intragenic polymorphisms and non-genetic factors. The main molecular mechanism underlying BH4 responsiveness is its chaperone-like effect on PAH, whereby it protects PAH protein integrity and rescues it from Ub-dependent degradation [99].
It is increasingly apparent that molecular chaperones could help mutant PAH proteins that are partially functional serve their purpose and help to prevent the pathogenic mechanisms that underlie genetic diseases.
Given the importance of chaperones, mutations in the chaperones themselves can be lethal. Multiple diseases are associated with mutations in the regulating chaperones. For example, a missense mutation in the equatorial domain of HSP60 causes spastic paraplegia, and mutation in tubulin-specific chaperone E causes hypoparathyroidism, mental retardation, and facial dysmorphism [100]. Similarly, in PKU an autosomal recessive mutation in DNAJC12, a PAH co-chaperone, reduced the activity of wild type PAH, leading to HPA. DNAJC12 is involved in PAH folding and interacts with the monoubiquitinated PAH variant, marking it for the Ub-dependent proteasomal/autophagy degradation system. Further studies are ongoing to elucidate the role of DNAJC12 in regulating PAH and PAH mutants [25,101]. Gene therapy and the ectopic expression of wild type chaperones might help to restore the partially functional mutant proteins [102,103].
Some patients with HT1 who are treated with NTBC (2-[2-nitro-4-(trifluoromethyl) benzoyl] cyclohexane-1,3-dione) also suffer from chronic hepatopathy and the development of hepatocellular carcinoma [104]. In a murine model of HT1, chaperones such as HSPB and HSPA were found to be associated with the anti-apoptotic proteins BCL-2 and BAG in the hepatocarcinogenetic process [105]. However, the role of molecular chaperones in FAH protein stability and degradation needs to be investigated.
Another system that can be targeted to rescue defective proteins is the UPS. As discussed in the previous section, the UPS is mainly driven by E1-E2-E3 enzymes that tag substrate proteins with ubiquitin molecules to mark them for degradation via the 26S proteasome, and DUBs can reverse that process. The role of DUBs in disease regulation has been imagined ever since their discovery, because they are involved in almost all cellular processes [106]. In PQC, the ubiquitin-mediated proteolytic pathway is a dynamic system responsible for regulating the fate of many proteins. In loss-of-function diseases, saving functional misfolded proteins from degradation can be a better alternative than dealing with a deficiency of proteins caused by rapid degradation. DUBs can rescue proteins from degradation by cleaving their degradative signals. Thus, DUBs act as proofreaders for mis-tagged substrate proteins and prevent them from degradation. In that way, DUBs could be used to curb protein misfolding diseases. It is unsurprising that direct evidence on this point is sparse. Most studies dealing with diseases related to protein folding problems aim to clear the misfolded proteins from the cells rapidly, and thus they target DUBs or the proteasome via specific inhibitors to prevent the pathogenesis of defective protein accumulation [107,108]. However, in diseases such as PKU and HT1, artificial manipulation of those systems could prove advantageous and pave the way for new therapeutic approaches. Nonetheless, the regulation of the proteostasis is not possible for those missense mutations which are present at the active site of the enzyme and other mutations causing truncation and splice variant. Therefore, controlling the proteostasis might be favorable only to the missense mutations that are located outside the active site.
PAH and FAH enzyme proteins are ubiquitinated and degraded by the Ub-dependent system, and therefore PAH and FAH mutants with high residual enzyme activity could be deubiquitinated by DUBs, which might suffice to create an adequate supply of functional protein. However, the mutations in certain genotypes can show dramatically different disease severities. Thus, in targeting DUBs as therapeutics for diseases with misfolded protein, it is important to understand the genotype-phenotype correlation and the allelic combination of mutations present in the genotype. A great deal of work remains to be done to improve understanding of how DUBs, molecular chaperones, and their combination can help to regulate enzyme deficiencies.


