 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ada Hang-Heng Wong | + 2255 word(s) | 2255 | 2020-08-14 04:48:54 | | | |
| 2 | Bruce Ren | Meta information modification | 2255 | 2020-08-24 10:33:12 | | | | |
| 3 | Bruce Ren | -21 word(s) | 2234 | 2020-10-27 10:40:15 | | |
Video Upload Options
Multiple myeloma (MM) is the second most common hematologic malignancy in the world. Even though survival rates have significantly risen over the past years, MM remains incurable, and is also far from reaching the point of being managed as a chronic disease.
1. Background
Multiple myeloma (MM) is the second most common hematologic malignancy, accounting for 1% of all cancers globally in 2016 . MM mainly occurs in older people, with a median age of 66–70 years at the time of diagnosis [1]. Age demography significantly contributes to MM progression and regimen design [2][3]. Survival rates are higher in younger people, partially due to the feasibility of autologous stem cell transplant (ASCT) and better drug tolerability during adjuvant therapy to ASCT or non-transplant systemic therapy. Nevertheless, overall survival rates have risen significantly over the past decades [4][5], with a five-year survival rate of 53.9% between 2010 and 2016 in the United States (US) [5]. Improved outcomes resulted from a significant increase of the use of novel therapies, including proteasome inhibitors and immunomodulatory drugs (IMiDs), which increased from 8.7% in 2000 to 61.3% in 2014 in US patients [6]. Additionally, the introduction of low-dose continuous therapy and maintenance therapy schemes post-ASCT or high-dose induction therapy also contributed to improved clinical outcomes, mainly through palliating adverse effects to increase drug tolerability, especially in weak elderly patients [7]. Aside from improvements in therapeutic design, novel drugs are designed to target specific molecular mechanisms involved in MM, especially the nuclear factor kappa B (NF-kB) signaling pathway [8][9][10], which is described in the following context.
2. Multiple Myeloma and NF-kB Signaling
Myeloma, also known as plasma cell myeloma, is the accumulation of malignant plasma cells in the bone marrow. Initially, in the asymptomatic phase (known as smoldering myeloma), myeloma cells produce abnormally substantial amounts of monoclonal proteins (M-proteins) that are released to the blood stream. As the disease progresses to the symptomatic phase, known as MM, the myeloma cells harness the bone marrow microenvironment to promote growth and invasion. MM progression leads to bone destruction, hypercalcemia and renal insufficiency, and may result in patient lethality.
MM is linked to the frequent onset of hyperploidy, chromosomal aberrations, genetic mutations and epigenetic transformations. Among these anomalies, hyperploidy is most prevalent, contributing to nearly half of all MM cases [11]. In non-hyperploid MM, chromosomal aberrations at the B cell class switching gene locus IGH frequently occur, leading to aberrant production of M-proteins in MM patients [12][13]. Aside from chromosomal changes, genetic mutations common in cancers frequently occur in MM too, e.g., the oncogenic KRAS and NRAS transformations and loss-of-function TP53 mutations [14][15][16]. Some genetic modifications are more MM-specific and contribute to hyperactive NF-kB signaling. These include the amplification or rearrangement of NIK, LTBR, TACI, NFKB1, NFKB2 and CD40 genes, as well as deletion or loss-of-function mutations in genes like CYLD, BIRC2/BIRC3 (cIAP1/cIAP2), TRAF2 and TRAF3 [17]. On the other hand, MM progression displays distinct epigenetic landscape changes. For example, extensive DNA hypomethylation in non-CpG islands occurs during the transition from monoclonal gammopathy of undetermined significance to the myeloma stage [18]. Moreover, hypermethylation of a subset of transcription factors, e.g., FOXD2, GATA4, RUNX2, and cell cycle-related genes, e.g., CDKN2B, potentially remodels cellular processes to promote tumorigenesis [18]. Furthermore, MM development is supported by the promoter methylation of the P53 gene, which is sustained by the NF-kB-regulated cytokine interleukin-6 (IL-6) [19]. Aside from DNA methylation, histone modifications such as acetylation and methylation also significantly alter the epigenetic landscape and drug response of MM [20]. For instance, overexpression of the histone methyltransferase gene EZH2 that frequently occurs in MM may be induced by hyperactive non-canonical NF-kB signaling [21]. Inhibition of EZH2 sensitizes MM to bortezomib treatment in vivo, through cooperative MYC suppression and inhibition of H3K27 trimethylation to regulate genes involved in B cell metabolism and antibody production [22][23]. NF-kB gene mutations are known to be the most prevalent in MM among all human cancers [24][25], and plays a pivotal role in anti-cancer therapy and drug resistance [26][27][28][29].
NF-kB refers to a family of transcription factors that form homo- and hetero-dimers within the family, as well as with other transcription factors [30]. NF-kB signaling is classified into the canonical and non-canonical pathways that are represented by the transcriptional protein complexes of p50/RelA and p52/RelB, respectively [31]. These two pathways are activated by distinct membrane receptors that respond to extracellular ligands like tumor necrosis factor a (TNFa), interleukin-1 (IL-1), receptor activator of NF-kB ligand (RANKL), and so on (Figure 1). In canonical NF-kB signaling, receptor activation leads to formation of the TRAF2-TRAF5-TRAF6 complex, which activates TAK1 kinase to phosphorylate the complex of IKKa, IKKb and NEMO. IKK complex phosphorylation subsequently triggers the degradation of IkB to release the p105 protein for proteasomal processing to the p50 protein. Consequently, the p50/RelA complex translocates to the nucleus and initiates transcription. Non-canonical NF-kB signaling involves the TRAF2-TRAF3-TRAF6 complex, which activates the NIK kinase to phosphorylate the IKKa kinase. Phosphorylated IKKa then triggers proteasomal processing of p100 to p52 for transcriptional activation. Although the canonical and non-canonical pathways have variant triggering signals and downstream targets, both pathways are involved in MM pathogenesis and progression .
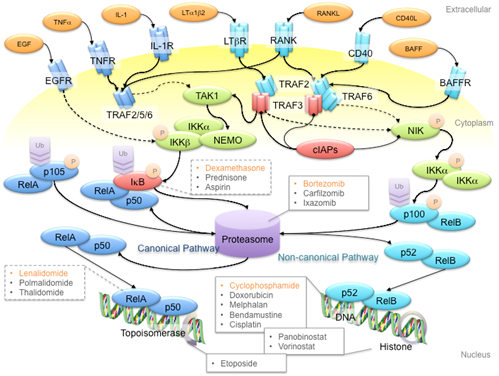
Figure 1. Schematic diagram of the NF-kB signaling pathway and anti-multiple myeloma (MM) drug targets. First-line anti-MM drugs (highlighted in orange) passively target the canonical and/or non-canonical pathways to shut down NF-kB signaling. For example, bortezomib inhibits the 26S proteasome to hinder the processing of p105 and p100 proteins, to prevent gene transcription activation in canonical and non-canonical NF-kB signaling pathways, respectively; dexamethasone induces IkB protein synthesis to inhibit p105 processing; lenalidomide reduces RelA binding to open chromatin; cyclophosphamide is a DNA alkylating agent that disrupts DNA replication and genome stability. Ligands, adaptor proteins and transcriptional complexes involved in canonical NF-kB signaling are depicted in dark blue, whereas those involved in non-canonical NF-kB signaling are depicted in light blue; kinases are depicted in green; inhibitors are depicted in red. P and Ub indicate the post-translational modifications of phosphorylation and ubiquitination, respectively. Arrows with triangle heads indicate activation, whereas arrows with rhomboid heads indicate inactivation/inhibition; direct interactions are indicated by solid lines, whereas indirect interactions are indicated by dash lines.
NF-kB signaling plays a pivotal role in promoting cancer growth, angiogenesis and tumor-microenvironment crosstalk, which mainly involves the production of pro-inflammatory cytokines, inflammation mediators, cell adhesion molecules, among others, to establish a favorable tumor microenvironment for MM tumorigenesis and disease progression. Non-canonical NF-kB signaling is also a key determinant of other oncogenic drivers, such as telomerase and telomeric proteins, which are commonly deregulated in cancers [32][33][34][35]. NF-kB signaling, in combination with other potent transcription factors such as STAT3, also plays important roles in regulating apoptosis and polarization of immune subtypes, which contribute to a pro-tumoral microenvironment [36][37]. Hence, many first-line anti-MM drugs have an indirect impact on the NF-kB signaling pathway (Figure 1). For instance, bortezomib is a reversible inhibitor of the 26S proteasome [38] and thus prevents the proteasomal cleavage of NF-kB proteins and the IkB protein to inhibit gene transcription activation. The insult of bortezomib on MM cells is further enhanced by the fact that the proteasome is overloaded by excessive M-protein production in myeloma cells. On the other hand, the corticosteroid dexamethasone induces inhibitor of kB (IkB) protein synthesis to inhibit NF-kB signaling [39]. Another first-line therapy drug, the IMiD lenalidomide, diminishes sustained RelA binding to open chromatin by inhibiting the Ikaros proteins Ikzf1 and Ikzf3 [40][41][42]. Co-administration of lenalidomide and dexamethasone suppresses interleukin-2 (IL-2), immunoglobin M (IgM) and immunoglobin G (IgG) production, hence reducing the protein load of MM patients [43]. In recent years, immunotherapy using antibodies and chimeric antigen receptor T (CAR-T) cells against specific NF-kB signaling receptors has gained more attention. For example, antibodies against B cell-activating factor (BAFF) inactivate non-canonical NF-kB signaling in MM cells [44][45]. Clinical trials are ongoing, so it is still too early to conclude whether any of these strategies works.
In addition to intracellular NF-kB hyperactivation, MM cells also manipulate NF-kB signaling in the bone marrow microenvironment to promote cancer growth and invasion (Figure 2). On the one hand, myeloma cells hijack stromal cells to secrete cytokines, such as interleukin-6 (IL-6), receptor activator of NF-kB ligand (RANKL) and vascular endothelial growth factor (VEGF), to promote cancer proliferation and angiogenesis [46]. On the other hand, myeloma cells secrete Dickkopf-1 (Dkk1) and macrophage inflammatory factor 1a (MIP-1a) to inhibit osteoblast differentiation to block new bone formation, and activate osteoclasts to promote osteolysis [47][48]. In late-stage MM patients, the myeloma cells acquire additional genetic abnormalities that lead to reduced dependency on the microenvironment (e.g., P53 mutation), increased drug resistance and increased aggressiveness of the clone (e.g., 1q21 amplification and CKS1B overexpression) [49]. Hence, therapies against both MM cells and microenvironment control, such as daratumab, an antibody drug against CD38 that induces antibody-dependent cytotoxic events in CD38-expressing cancer cells and complement-dependent cytotoxicity [50][51][52], show success in MM treatment [53].
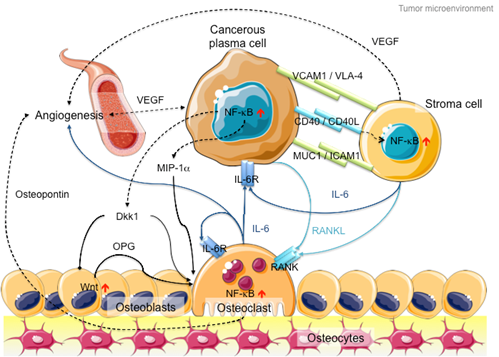
Figure 2. Schematic diagram of the MM microenvironment. Cancerous plasma cells interact with stroma, osteoblasts and osteoclasts through membrane receptor interactions and secretory cytokine pathways. Hyperactive NF-kB signaling plays a pivotal role in disease progression through transcriptional activation of the secretion of various cytokines like IL-6, RANKL and Dkk1 to promote cancer cell proliferation, osteoblast inactivation, osteoclast hyperactivation and angiogenesis. Proteins involved in canonical NF-kB signaling are indicated in dark blue, whereas proteins involved in non-canonical NF-kB signaling are indicated in light blue. Arrows with triangle heads indicate activation, whereas arrows with rhomboid heads indicate inactivation/inhibition; direct interactions are indicated by solid lines, whereas indirect interactions are indicated by dashed lines. Artistic images were downloaded from Servier Medical Art (https://smart.servier.com/; Servier Medical Art by servier is licensed under a creative commons attribution 3.0 unported license).
Even though receptor-/ligand-specific antibodies can target NF-kB signaling with high specificity, the diverse NF-kB signals in MM cells and their microenvironment limit the application of these therapies to treat MM effectively in vivo. Tumor evolution also contributes to altering pathways to develop drug resistance. Hence, directly targeting the molecular machinery of NF-kB signaling remains crucial. Until now, no specific NF-kB inhibitor has been approved for treating MM. We will discuss the challenges of developing specific NF-kB inhibitors for MM treatment.
3. NF-kB Signaling: The Rose with Thorns in MM Treatment
Even though NF-kB signaling plays a critical role in MM, specifically targeting this signaling pathway proves to be more difficult than previously thought.
The foremost hurdle is drug safety. NF-kB signaling plays a key role in innate immunity and inflammation. Constitutive inactivation of NF-kB signaling silences the immune system, subsequently rendering patients susceptible to infections. Population-based studies have pointed out that MM patients displayed a seven-fold higher risk of bacterial infection and a 10-fold higher risk of viral infections as compared to randomized control individuals, resulting in a stunning 22% death rate among MM patients at one-year follow-up [54]. It is noteworthy that transplanted patients displayed a broader spectrum of infection [55], where infection rate may be reduced by combined IMiD therapy [56], due to the fact that immunosuppressive drugs are administered to prevent graft-versus-host defense. On the other hand, bortezomib-based therapy is associated with a higher risk of severe infection in various studies [56][57][58], possibly through inhibition of both canonical and non-canonical NF-kB pathways. Hence, it is hypothesized that targeting of one NF-kB pathway may be safer than inactivating both NF-kB signaling pathways. Nevertheless, trials of many IKKb inhibitors showed severe adverse effects [59], even though non-canonical NF-kB signaling is hypothesized to be unaffected. In contrast, the anti-RANK antibody denosumab, which is recommended as adjuvant therapy to MM patients to alleviate hypercalcemia due to hyperactive osteoclasts [60], produces little toxicity but cannot treat MM because of its narrow-spectrum inhibition of RANK-mediated non-canonical NF-kB signaling. Hence, striking the right balance between drug safety and treatment efficiency remains challenging.
Secondly, the context-specific and spatio-temporal regulation of NF-kB signaling complicates therapeutic design. This complexity is further complicated by the interplay between cancer and its immune microenvironment [61][62]. For example, the non-steroidal anti-inflammatory drugs (NSAIDs) (e.g., aspirin, sulindac and tolfenamic acid) that target the COX-1/2 genes may suppress NF-kB signaling during short-term administration but activate NF-kB signaling after prolonged treatment [59].
Thirdly, selectivity remains a critical issue. For instance, several IKKb inhibitors exhibit off-target effects, whereas others display high selectivity toward IKKb and loss of inhibition through IKKa [59]. In this regard, combined treatment may offer hope for highly selective inhibitors, but this also requires more consideration of the treatment burden on patients, especially when many MM patients are old and weak. Nevertheless, high selectivity suffers from poor treatment efficiency. For instance, the anti-BAFF antibody tabalumab, which specifically inhibits non-canonical NF-kB signaling, failed to improve outcomes in MM patients in a phase II trial in which it was combined with bortezomib and dexamethasone [63].
Lastly, the lack of structural information hampers rational drug design, although solving the structure does not guarantee success either. For example, structure of the NF-kB inducing kinase (NIK) has been solved [64], but the NIK inhibitors AM-0216 and AM-0561 failed due to poor pharmacokinetics in vivo, even though they displayed high NIK binding affinity and NIK-dependent cytotoxicity in vitro [65]. On the contrary, a peptide mimetic of the NF-kB essential modulator (NEMO) binding domain that blocks IKK complex formation [66] made its way to clinical trials in dogs for treating diffuse large B cell lymphoma (DLBCL) and soft tissue sarcoma (STS) [67], but no clinical trial has been reported to treat humans. Other molecular targets might lack specific inhibitors after solving the structure [68]. Consequently, no specific NF-kB inhibitor has been successfully developed to treat MM until now.
References
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. In Seminars in Oncology; W.B. Saunders (United States): 2016; pp. 676–681.
- Palumbo, A.; Bringhen, S.; Ludwig, H.; Dimopoulos, M.A.; Bladé, J.; Mateos, M.V.; Rosiñol, L.; Boccadoro, M.; Cavo, M.; Lokhorst, H.; et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: A report of the European Myeloma Network (EMN). Blood 2011, 118, 4519–4529.
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060.
- Thorsteinsdottir, S.; Dickman, P.W.; Landgren, O.; Blimark, C.; Hultcrantz, M.; Turesson, I.; Björkholm, M.; Kristinsson, S.Y. Dramatically improved survival in multiple myeloma patients in the recent decade: Results from a Swedish population-based study. Haematologica 2018, 103, e412–e415.
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review (1975–2017). Bethesda, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 3 May 2020).
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000-2014. Leukemia 2017, 31, 1915–1921.
- Dimopoulos, M.A.; Jakubowiak, A.J.; McCarthy, P.L.; Orlowski, R.Z.; Attal, M.; Bladé, J.; Goldschmidt, H.; Weisel, K.C.; Ramasamy, K.; Zweegman, S.; et al. Developments in continuous therapy and maintenance treatment approaches for patients with newly diagnosed multiple myeloma. Blood Cancer J. 2020, 10, 17.
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429.
- Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the involvement of the master transcription factor NF-κB in cancer initiation and progression. Biomedicines 2018, 6, 82, doi:10.3390/biomedicines6030082.
- Chew, C.L.; Conos, S.A.; Unal, B.; Tergaonkar, V. Noncoding RNAs: Master Regulators of Inflammatory Signaling. Trends Mol. Med. 2018, 24, 66–84.
- Chesi, M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma: Basic and clinical updates. Int. J. Hematol. 2013, 97, 313–323.
- Bergsagel, P.L.; Nardini, E.; Brents, L.; Chesi, M.; Kuehl, W.M. IgH translocations in multiple myeloma: A nearly universal event that rarely involves c-myc. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 1997; pp. 283–287.
- Fonseca, R.; Debes-Marun, C.S.; Picken, E.B.; Dewald, G.W.; Bryant, S.C.; Winkler, J.M.; Blood, E.; Oken, M.; Santana-Dávila, R.; González-Paz, N.; et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood 2003, 102, 2562–2567.
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101.
- Demchenko, Y.N.; Michael Kuehl, W. A critical role for the NFκB pathway in multiple myeloma. Oncotarget 2010, 1, 59–68.
- Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells 2020, 9, 287.
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell 2007, 12, 131–144.
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562.
- Hodge, D.R.; Peng, B.; Cherry, J.C.; Hurt, E.M.; Fox, S.D.; Kelley, J.A.; Munroe, D.J.; Farrar, W.L. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res. 2005, 65, 4673–4682.
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The epigenome in multiple myeloma: Impact on tumor cell plasticity and drug response. Front. Oncol. 2018, 8, 566.
- Iannetti, A.; Ledoux, A.C.; Tudhope, S.J.; Sellier, H.; Zhao, B.; Mowla, S.; Moore, A.; Hummerich, H.; Gewurz, B.E.; Cockell, S.J.; et al. Regulation of p53 and Rb Links the Alternative NF-κB Pathway to EZH2 Expression and Cell Senescence. PLoS Genet. 2014, 10, e1004642.
- Rizq, O.; Mimura, N.; Oshima, M.; Saraya, A.; Koide, S.; Kato, Y.; Aoyama, K.; Nakajima-Takagi, Y.; Wang, C.; Chiba, T.; et al. Dual Inhibition of EZH2 and EZH1 Sensitizes PRC2-Dependent Tumors to Proteasome Inhibition. Clin. Cancer Res. 2017, 23, 4817–4830, doi:10.1158/1078-0432.CCR-16-2735.
- Guo, M.; Price, M.J.; Patterson, D.G.; Barwick, B.G.; Haines, R.R.; Kania, A.K.; Bradley, J.E.; Randall, T.D.; Boss, J.M.; Scharer, C.D. EZH2 Represses the B Cell Transcriptional Program and Regulates Antibody-Secreting Cell Metabolism and Antibody Production. J. Immunol. 2018, 200, 1039–1052.
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent Engagement of the Classical and Alternative NF-κB Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell 2007, 12, 115–130.
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Leif Bergsagel, P.; Michael Kuehl, W. Classical and/or alternative NF-κB pathway activation in multiple myeloma. Blood 2010, 115, 3541–3552.
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two sides of the same coin. Genes (Basel) 2018, 9, 24, doi:10.3390/genes9010024.
- Eluard, B.; Thieblemont, C.; Baud, V. NF-κB in the New Era of Cancer Therapy. Trends Cancer 2020, 6, 677–687, doi:10.1016/j.trecan.2020.04.003.
- Dehghanifard, A.; Kaviani, S.; Abroun, S.; Mehdizadeh, M.; Saiedi, S.; Maali, A.; Ghaffari, S.; Azad, M. Various Signaling Pathways in Multiple Myeloma Cells and Effects of Treatment on These Pathways. Clin. Lymphoma Myeloma Leuk. 2018, 18, 311–320.
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57.
- Shih, V.F.S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102.
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830.
- Li, Y.; Cheng, H.S.; Chng, W.J.; Tergaonkar, V.; Cleaver, J.E. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc. Natl. Acad. Sci. USA 2016, 113, 14402–14407.
- Akincilar, S.C.; Low, K.C.; Liu, C.Y.; Yan, T.D.; Oji, A.; Ikawa, M.; Li, S.; Tergaonkar, V. Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett. 2015, 589, 974–984.
- Khattar, E.; Maung, K.Z.Y.; Chew, C.L.; Ghosh, A.; Mok, M.M.H.; Lee, P.; Zhang, J.; Chor, W.H.J.; Cildir, G.; Wang, C.Q.; et al. Rap1 regulates hematopoietic stem cell survival and affects oncogenesis and response to chemotherapy. Nat. Commun. 2019, 10, 1–14.
- Ozturk, M.B.; Li, Y.; Tergaonkar, V. Current insights to regulation and role of telomerase in human diseases. Antioxidants 2017, 6, 17, doi:10.3390/antiox6010017.
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506.
- Siveen, K.S.; Nguyen, A.H.; Lee, J.H.; Li, F.; Singh, S.S.; Kumar, A.P.; Low, G.; Jha, S.; Tergaonkar, V.; Ahn, K.; et al. Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. Br. J. Cancer 2014, 111, 1327–1337.
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; P. Dou, Q. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253.
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-κB activity through induction of IκB synthesis. Science 1995, 270, 286–290.
- Oh, K.-S.; Gottschalk, R.A.; Lounsbury, N.W.; Sun, J.; Dorrington, M.; Baek, S.; Sun, G.; Wang, Z.; Krauss, K.S.; Milner, J.D.; et al. Dual Roles for Ikaros in Regulation of Macrophage Chromatin State and Inflammatory Gene Expression. J. Immunol. 2018, 201, 757–771.
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305.
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 2014, 343, 305–309.
- Shannon, E.; Sandoval, F.; Greig, N.; Stagg, P. Lenalidomide alone or lenalidomide plus dexamethasone significantly inhibit IgG and IgM in vitro⋯A possible explanation for their mechanism of action in treating multiple myeloma. Int. Immunopharmacol. 2012, 12, 441–446.
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.-F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157.
- Tai, Y.T.; Li, X.F.; Breitkreutz, I.; Song, W.; Neri, P.; Catley, L.; Podar, K.; Hideshima, T.; Chauhan, D.; Raje, N.; et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006, 66, 6675–6682.
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget 2017, 8, 20394–20409.
- Pinzone, J.J.; Hall, B.M.; Thudi, N.K.; Vonau, M.; Qiang, Y.-W.; Rosol, T.J.; Shaughnessy, J.D. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009, 113, 517–525.
- Hata, H. Bone lesions and macrophage inflammatory protein-1 alpha (MIP-1α) in human multiple myeloma. Leuk. Lymphoma 2005, 46, 967–972.
- Chng, W.J.; Glebov, O.; Bergsagel, P.L.; Kuehl, W.M. Genetic events in the pathogenesis of multiple myeloma. Best Pract. Res. Clin. Haematol. 2007, 20, 571–596.
- de Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848.
- Kaku, H.; Horikawa, K.; Obata, Y.; Kato, I.; Okamoto, H.; Sakaguchi, N.; Gerondakis, S.; Takatsu, K. NF-kB is required for CD38-mediated induction of Cg1 germline transcripts in murine B. lymphocytes. Int. Immunol. 2002, 14, 1055–1064.
- Qian, Y.; Chen, C.; Ma, L.; Wang, Z.; Wang, L.-F.; Zuo, L.; Yang, Y.; Huang, X.; Jiang, M.; Wang, X.; et al. CD38 Deficiency Promotes Inflammatory Response Through Activating Sirt1/NF-κB-Mediated Inhibition of TLR2 Expression in Macrophages. Mediat. Inflamm. 2018, 2018, 1–13, doi:10.1155/2018/8736949.
- Neri, P.; Kumar, S.; Fulciniti, M.T.; Vallet, S.; Chhetri, S.; Mukherjee, S.; Tai, Y.; Chauhan, D.; Tassone, P.; Venuta, S.; et al. Neutralizing B-cell-activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin. Cancer Res. 2007, 13, 5903–5909.
- Blimark, C.; Holmberg, E.; Mellqvist, U.H.; Landgren, O.; Björkholm, M.; Hultcrantz, M.; Kjellander, C.; Turesson, I.; Kristinsson, S.Y. Multiple myeloma and infections: A population-based study on 9253 multiple myeloma patients. Haematologica 2015, 100, 107–113.
- Nucci, M.; Anaissie, E. Infections in Patients with Multiple Myeloma in the Era of High-Dose Therapy and Novel Agents. Clin. Infect. Dis. 2009, 49, 1211–1225.
- Teh, B.W.; Harrison, S.J.; Worth, L.J.; Thursky, K.A.; Slavin, M.A. Infection risk with immunomodulatory and proteasome inhibitor–based therapies across treatment phases for multiple myeloma: A systematic review and meta-analysis. Eur. J. Cancer 2016, 67, 21–37.
- Li, J.; Li, Y.; Huang, B.; Zheng, D.; Chen, M.; Zhou, Z. Drug-Induced Modulation of T Lymphocytes as a Potential Mechanism of Susceptibility to Infections in Patients with Multiple Myeloma During Bortezomib Therapy. Cell Biochem. Biophys. 2014, 71, 457–464.
- Offidani, M.; Corvatta, L.; Bringhen, S.; Gentili, S.; Gay, F.; Maracci, L.; Boccadoro, M.; Leoni, P.; Palumbo, A. Infection Complications in 476 Patients with Newly Diagnosed Multiple Myeloma Treated with Lenalidomide or Bortezomib Combinations. Blood 2015, 126, 5365–5365.
- Prescott, J.; Cook, S. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115.
- Goldstein, D.A. Denosumab for bone lesions in multiple myeloma—What is its value? Haematologica 2018, 103, 753–754.
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324.
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723.
- Raje, N.S.; Moreau, P.; Terpos, E.; Benboubker, L.; Grząśko, N.; Holstein, S.A.; Oriol, A.; Huang, S.-Y.; Beksac, M.; Kuliczkowski, K.; et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br. J. Haematol. 2017, 176, 783–795.
- Liu, J.; Sudom, A.; Min, X.; Cao, Z.; Gao, X.; Ayres, M.; Lee, F.; Cao, P.; Johnstone, S.; Plotnikova, O.; et al. Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 2012, 287, 27326–27334.
- Demchenko, Y.N.; Brents, L.A.; Li, Z.; Bergsagel, L.P.; McGee, L.R.; Kuehl, M.W. Novel inhibitors are cytotoxic for myeloma cells with NFkB inducing kinase-dependent activation of NFkB. Oncotarget 2014, 5, 4554–4566.
- Rushe, M.; Silvian, L.; Bixler, S.; Chen, L.L.; Cheung, A.; Bowes, S.; Cuervo, H.; Berkowitz, S.; Zheng, T.; Guckian, K.; et al. Structure of a NEMO/IKK-Associating Domain Reveals Architecture of the Interaction Site. Structure 2008, 16, 798–808.
- Habineza Ndikuyeze, G.; Gaurnier-Hausser, A.; Patel, R.; Baldwin, A.S.; May, M.J.; Flood, P.; Krick, E.; Propert, K.J.; Mason, N.J. A Phase I Clinical Trial of Systemically Delivered NEMO Binding Domain Peptide in Dogs with Spontaneous Activated B-Cell like Diffuse Large B-Cell Lymphoma. PLoS ONE 2014, 9, e95404.
- Xu, X.; Li, Y.; Bharath, S.R.; Ozturk, M.B.; Bowler, M.W.; Loo, B.Z.L.; Tergaonkar, V.; Song, H. Structural basis for reactivating the mutant TERT promoter by cooperative binding of p52 and ETS1. Nat. Commun. 2018, 9, 3138, doi:10.1038/s41467-018-05644-0.


