 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francisco Aguayo | + 3880 word(s) | 3880 | 2020-08-11 11:42:46 | | | |
| 2 | Bruce Ren | -20 word(s) | 3860 | 2020-08-12 05:39:23 | | | | |
| 3 | Bruce Ren | Meta information modification | 3860 | 2020-08-21 11:44:39 | | | | |
| 4 | Bruce Ren | Meta information modification | 3860 | 2020-08-21 12:21:30 | | |
Video Upload Options
Human papillomavirus (HPV) infection is not a sufficient condition for cervical carcinogenesis. Tobacco smoke is a recognized cofactor. In this manuscript, we described the molecular mechanisms by which tobacco smoke affects the HPV role in cervical cancer.
Cervical, anogenital, and some head and neck cancers (HNC) are etiologically associated with high-risk human papillomavirus (HR-HPV) infection, even though additional cofactors are necessary. Epidemiological studies have established that tobacco smoke (TS) is a cofactor for cervical carcinogenesis because women who smoke are more susceptible to cervical cancer when compared to non-smokers. Even though such a relationship has not been established in HPV-related HNC, a group of HPV positive patients with this malignancy are smokers. TS is a complex mixture of more than 4500 chemical compounds and approximately 60 of them show oncogenic properties such as benzo[α]pyrene (BaP) and nitrosamines, among others. Some of these compounds have been evaluated for carcinogenesis through experimental settings in collaboration with HR-HPV. Here, we conducted a comprehensive review of the suggested molecular mechanisms involved in cooperation with both HR-HPV and TS for epithelial carcinogenesis. Furthermore, we propose interaction models in which TS collaborates with HR-HPV to promote epithelial cancer initiation, promotion, and progression. More studies are warranted to clarify interactions between oncogenic viruses and chemical or physical environmental factors for epithelial carcinogenesis.
1. Introduction
Cervical cancer is a malignant neoplasia that develops in the cervix, the lower part of the uterus that connects this with the vagina [1]. This disease is initiated through a precursor epithelial lesion with slow and progressive evolution, known as cervical intraepithelial neoplasia (CIN I, II, and III). When only stratified epithelium is affected, it constitutes an in-situ cervical carcinoma. Eventually, the progression to invasive cancer occurs when tumor cells invade additional tissues [2]. Human papillomavirus (HPV) infection is the necessary condition for developing cervical carcinoma, because almost 100% of patients with cervical cancer are positive for HPV. However, a low percentage of HPV infected women ultimately develop low-grade CIN. Moreover, a low percentage of them progress towards high-grade CIN and invasive cervical carcinoma. Despite this, cervical cancer is an important public health problem, since 311,000 deaths per year are directly associated with this neoplasia [3]. In addition, 80% of these deaths occur in developing countries. High-risk zones of cervical cancer are South America, sub-Saharan Africa, and India. In the United States, there are more than 29,000 new cases per year including more than 11,000 deaths. The mortality rates are especially high in Chile and Mexico, while a low mortality rate is observed in Cuba, Puerto Rico, and Argentina [4][5]. On the other hand, head and neck cancer (HNC) is a heterogeneous group of tumors that arise in the head and neck region, which include the oral cavity, pharynx (e.g., nasopharynx, oropharynx, hypopharynx), paranasal sinuses, nasal cavity, larynx, and salivary glands [6]. HNCs are the sixth most frequent form of cancers worldwide, with an annual incidence of over 500,000 new cases . Tobacco smoke (TS), alcohol consumption, and some viral infections, among other factors, are involved in its development [7]. HPV infection, in particular, has been etiologically associated with a subset of oropharyngeal and oral carcinomas [8]. When considering all HNCs, approximately 25% constitute HPV positive tumors, with a higher prevalence in oropharyngeal cancer (35.6%, range 11–100%) than in the oral cavity (23.5%, range 40–80%) or larynx (24%, range 0–100%) [9]. HPV is the main causal agent of the increased incidence of squamous cell carcinoma (SCC) of the oropharynx in developed countries, especially in males. The disease has a tendency to affect younger patients compared to those with HPV negative oropharyngeal carcinoma [10]. Epidemiologically, it is accepted that HPV positive oropharyngeal carcinomas are entities with a differential clinical behavior when compared to HPV negative cases [11].
2. Tobacco Smoke and HPV Interactions
2.1. History and Epidemiology
As previously stated, additional factors are necessary for HPV-mediated carcinogenesis [12]. It has been proposed that some environmental chemical compounds alter HPV gene expression. In fact, in the first half of the 20th century, Peyton Rous discovered a cooperation between tar and papillomaviruses for inducing rabbit SCCs [13]. These historical experiments demonstrated that carcinogenesis may be induced by exposure to two different factors. In 1977, Winkelstein hypothesized that SCCs from different sites will be associated with TS. Therefore, considering that most cervical cancers are SCCs, it is expected that this tumor will be associated with TS [14]. In 1993, it was reported a possible cooperation between HPV16 and tobacco-related carcinogens for oral carcinogenesis. The authors demonstrated that primary oral keratinocytes immortalized with HPV16 acquire a tumor phenotype after treatment with nitrosamines and nitroguanosine, two carcinogens present in TS [15].
Epidemiological evidence strongly suggests a synergy between TS and HR-HPV infection for cervical cancer development [16][17][18]. In 2003, the IARC established that women who smoke are more susceptible to cervical cancer when compared to nonsmokers [19]. However, the mechanisms involved in such collaboration are unclear. According to the World Health Organization (WHO), TS is considered the most important human carcinogen involved in cancer initiation, promotion, and progression. Additionally, TS is considered the major cause of morbidity and mortality worldwide, causing more than five million deaths each year [20]. TS is a complex mixture of more than 4500 chemical compounds such as carbon monoxide (CO), hydrogen cyanide (HCN), nitrogen oxides (NOX), formaldehyde, acrolein, benzene, nicotine, nitrosamines, phenol/polycyclic aromatic hydrocarbons (PAH), and more [21]. Considering the established evidence that both TS and HPV are involved in epithelial cancer initiation, promotion, and progression, we speculate that a complex network of interactions exists. Due to the detection of TS metabolites in cervical mucous of women who smoke [22][23] and the direct exposure of oral cavity and nasopharynx to TS, the presence of both tobacco and HPV increases the possibility of direct interaction. Thus, considering molecular evidence, different findings have been reported (Table 1).
2.2. Tobacco Smoke Affects HPV Replication
Alam et al. demonstrated that benzo[α]pyrene (BaP), a carcinogen present in TS, is able to increase the number of virions and genomes of HPV31 in organotypic epithelial cultures of cervical cells. The authors observed that high BaP concentrations resulted in a 10-fold increase in viral titers while low BaP concentrations increase the number of viral genomes [24]. It was suggested that this effect may increase the possibility of viral dissemination and persistence. Later, the same authors demonstrated that the Ras-Raf-Mek1/2-Erk1/2 signaling pathway is involved in the increase of HPV31 virions after BaP exposure. Moreover, Erk1/2 signaling promoted activation of CDK1 [25]. However, epidemiological studies did not find a dose-response relationship between TS frequency and HPV DNA load in cervical cancer [26][27][28]. In head and neck cancer HPV positive cases, studies reporting a TS dose-dependence respect viral load are lacking. More studies are warranted to explore this possibility.
2.3. Tobacco Smoke Promotes HPV E6 and E7 Expression
One study suggested that nicotine, an addictive component which is contained at high levels in TS, enhances the activity of HPV16 LCR synergistically with Brn-3a transcription factor and consequently promotes the expression of E6 and E7 oncoproteins [29]. Additionally, a recent report demonstrated that BaP increases the expression of HPV16 E7 in organotypic epithelial cultures of cervical cells. This expression is inhibited by curcumin, a potential chemopreventive natural compound [30]. On the other hand, Wei et al. demonstrated that acute exposure to cigarette smoke extracts (CSE) allows an increase in E6 and E7 when cervical cells maintain HR-HPV in an episomal physical status [31]. In the same way, Peña et al. reported that CSE promote HPV16 p97 promoter activation in lung epithelial cells, resulting in increased E6/E7 levels [32]. The mechanism involved in TS-mediated E6 and E7 expression is unclear, although it seems that AP-1 may be involved [33]. The activity of AP-1 can be regulated by c-Jun and c-Fos phosphorylation through MAPK pathway activation in response to diverse stimuli (i.e., exposure to TS) [34]. Interestingly, it was reported that CSE enhances AP-1 activity, which in turn up-regulates proangiogenic cytokines in oropharyngeal cells [35]. In addition, carcinogens from TS promote mutations that favor AP-1 binding to HPV16 LCR, increasing the activity of p97 promoter [36][37]. Moreover, evidence indicates that TS promotes AP-1 activity in several cell types [38][39][40]. In lung cells, transcription factors, such as NF-κB, HIF, and AP-1, have been shown to be highly sensitive to CSE [41][42], playing a critical role in the inflammatory response due to injury induced by reactive oxygen species (ROS) [43]. In this scenario, AP-1 activity can be altered by the induction of kinases that respond to the dimerization and phosphorylation of tyrosine kinases receptors. In fact, in the TS-induced lung pathogenesis, the EGFR was found to be overexpressed and aberrantly activated by TS [44]. The classical pathway induced by TS is MAPK, which through multiple factors, mainly ERK1/2, can direct the signal to the nucleus to promote AP-1 phosphorylation, promoting their binding to cognate regulatory elements in cell promoters[45][46][47]. Moreover, TS has been demonstrated to induce upregulation of c-Jun, c-Fos, and Fra-1, but not of Fra-2, Jun-B, and Jun-D expression [48], suggesting an obligatory role for EGFR-mediated MAPK activation. Evidence also reveals that TS induces the expression of cyclin D1 and PCNA, which are AP-1 targets genes involved in promoting cell cycle progression [108]. Likewise, in human bronchial cells, it has been shown that TS induces increased AP-1 activity, promoting aberrant c-Jun/Fra1 dimerization, which is linked with aberrant phenotypical changes such as hyperplasia, epithelial-mesenchymal transition (EMT), release of EGF ligand, and cell transformation [49][50]. Additionally, AP-1 activity is regulated by TS at different levels through its phosphorylation, dimer composition, and mRNA expression [103]. Taken together, it is plausible that AP-1 is involved in TS-mediated E6 and E7 overexpression.
2.4. Tobacco Smoke Affects Immune Responses Against HPV
It has been established that TS is associated with early cervical carcinogenic alterations and a decreased ability of the immune system to induce HPV clearance in the cervix [51][52]. In fact, TS affects both innate and adaptive immune responses [53]. One study reported that NK cell activity was significantly decreased in subjects who smoked [54][55]. On the other hand, the same study reported a decreased amount of serum antibodies in subjects who smoked, explaining, at least in part, a higher susceptibility to infections. In addition, a reduction of Langerhans cells and helper T cells in the uterine cervix transformation zone for women who smoke was reported, suggesting a decreased local immunosurveillance [56]. Moreover, a reduction of smoking was associated with changes in immune cell counts [57]. It was further demonstrated that TS shows direct immunosuppressive effects on T cells, increasing the percentage of CD8+ T cells while lowering CD4+ T cells [58].
Regarding specific compounds, nicotine shows immunosuppressive properties in some animal models [59][60]. Moreover, nicotine is able to promote migration in cervical cancer cells by activating the PI3K/Akt/NF-κB pathway, which suggests connections between TS and cervical cancer progression [61]. Interestingly, acrolein, a major component of TS, it was reported to suppress the inflammatory and innate response by reducing levels of IL-8 mRNA and human beta-defensin (HBD) in sinonasal epithelial cells [62]. Alike, BaP, at low-dose, was demonstrated to have immunosuppressive effects in activated murine marrow-derived macrophages in an aryl hydrocarbon receptor (AhR)-dependent manner [63]. Taken together, studies support immunosuppressive effects of TS and/or some specific compounds such as nicotine, BaP, and acrolein, which may favor increased infection susceptibility and/or impairing its clearance.
2.5. Tobacco Smoke Promotes DNA Damage Leading to an Increased HPV Oncogenic Role
The most important molecular alterations caused by TS are adduct generation and the activation of signaling pathways after binding tobacco components (i.e., nicotine) to membrane receptors [64]. Some compounds present in TS are processed and metabolically activated by oxidoreductases, such as cytochrome P450, after which hydrosoluble electrophilic molecules bind to the genome leading the production of covalent and stable complexes with nitrogenated bases, known as DNA adducts [65][66] which can lead to mutations and cancer [67].
It has been demonstrated that HPV16 and 18 infected cells are capable of generating high levels of BaP metabolites, thus increasing adduct formation, DNA damage and probably, favoring the possibility of HPV integration into the host [68]. In accordance with this finding, HPV-transformed cervical cancer cells were highly susceptible to DNA damage promoted by CSE, detected by comet assay [69]. Interestingly, Wei et al. found increased DNA mutations and double strand breaks in CIN I cells harboring episomal HPV after TS exposure . Moreover, the authors showed that TS increases p53 levels in normal HPV-negative cervical cells, thus activating DNA repair and apoptosis. On the contrary, in HR-HPV-infected cells, TS induces a decrease in p53 activity by promoting overexpression of viral oncoproteins. The authors finally suggested that cervical cells harboring episomal HPV are more susceptible to DNA damage promoted by TS, when compared to those cells harboring integrated HPV, concluding a more pronounced effect in early lesions. In line with this finding, an article revealed higher levels of p53 in both cervical cancer cells and HPV (E6E7) - transfected cells exposed to TS. Additionally, p53 expression was not altered in HPV-negative (C33A) human cervical cells exposed to TS for 72 h. These findings suggest the presence of residual p53 activity in HPV positive cervical carcinoma cells [70][71].
It is known that TP53 is frequently mutated in HPV negative tumors, such as those associated with TS[72][73]. Specifically, missense TP53 mutations frequently result in p53 accumulation in the nucleus of tumor cells [74]. Regarding this issue, an in vivo study in head and neck carcinoma samples from 110 patients showed that tobacco consumption was highly associated with p53 staining intensity on immunohistochemistry [75]. On the contrary, TP53 is not frequently mutated in HPV-driven tumors, even though p53 levels are significantly decreased by HR-HPV E6 oncoprotein. Thus, in non-tumor HPV infected cells, TS exposure promotes functional p53 activity, which is abrogated by E6 oncoprotein, leading to increased DNA damage and cancer initiation . On the other hand, Muñoz et al. found that TS and E6/E7 collaborates for increasing tumor properties of lung epithelial cells [76]. Moreover, Peña et al. showed that E6 and E7 cooperate with TS for increasing DNA damage in tumor epithelial cells . Another study reported a higher sensitivity to the inactivation of clonogenic survival to BaP in HR-HPV E6, expressing fibroblasts compared to normal and inactive-p53 lines (p53-H179Q and p53-RNAi) exposed to the same cigarette smoke carcinogen. Moreover, it was proposed that HR-HPV E6 may activate G2 checkpoint kinase CHK1, a well-known DNA damage marker, in fibroblasts exposed to BaP. Altogether, all of these data suggest a synergistic interaction between TS and HPV for increasing the DNA damage in epithelial cells, ultimately leading to cancer initiation or progression (Figure 1) .
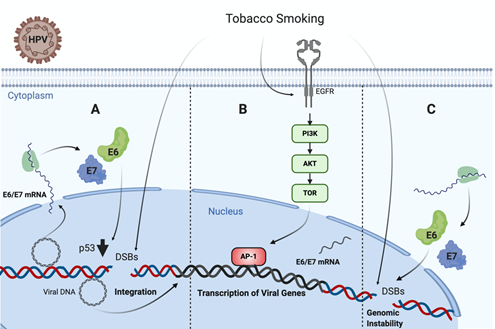
Figure 1. Tobacco smoke cooperates with high-risk human papillomavirus (HR-HPV) for increased DNA damage in epithelial cells. (A) Tobacco smoke causes E6/E7 oveerexpression and DNA damage in cells harboring HPV episomal forms, inducing p53 downregulation and potentially promoting viral genome integration [101]. (B) Tobacco smoke promotes EGFR/PI3K/AKT activation inducing AP-1 recruitment to the LCR and activating the HR-HPV early promoter, thus increasing E6/E7 expression. (C) Both tobacco smoke and E6/E7 oncoproteins cooperate for increasing genomic instability in infected cells .
2.6. Tobacco Smoke Alters Cellular Gene Expression Involved in HPV-Mediated Carcinogenesis
Due to the strong relationship between TS and lung cancer, the role of tobacco has been highly studied in lung cells and patients with this disease. In fact, it has been demonstrated that lung cells exposed to CSE show gene expression alterations in around 3700 genes, the cytochrome P450 enzyme among them . In lung cells, the exposure to CSE is associated with chromatin remodeling, which, in turn, leads to differential gene expression involved in oxidative stress responses. Interestingly, these effects are enhanced in patients with viral infections [77].
As previously mentioned, viral oncogenes E6 and E7 play a crucial role interfering with pRb and p53 tumor suppressor functions, which are important events in HPV-mediated carcinogenesis [78]. Some studies have assessed the interplay between TS and HPV in such gene expression. The effect of HPV and TS on p53 was addressed in Section 3.5. On the other hand, Alam et al. evaluated the effect of BaP on tumor suppressor pRb and other proteins expressions in HPV31b+ cervical intraepithelial neoplasia culture. They observed that inactive pRb (hyperphosphorylated) form was significantly upregulated compared to the negative control, proposing that the regulation by BaP may not be correlated with the HPV E7 effect. Interestingly, CDK1, a protein active in the G2 phase of the cell cycle, exhibited a significant increase in HPV31b raft cultures treated with BaP. The authors suggested that this carcinogenic agent may modulate CDK1 kinase activity in HPV-positive in order to favor cancer progression [79]. This may be supported by the evidence that E6 could upregulate CDK1 through E2F1, shedding light on a promising mechanism in which HPV induces genomic instability [80].
Another study demonstrated that eukaryotic translation initiation factor 4E (eIF4E), a crucial factor in translation control and recently evaluated as a plausible oncogene, is induced by E6 and E7 proteins. It has been shown to influence cell proliferation, migration, and apoptosis [81][82]. Moreover, the contribution between nicotine and HPV was assessed in another study. This article revealed that nicotine upregulates eIF4E expression in HPV-immortalized cervical epithelial (H8) cells, and subsequently enhances the expression of oncogenes c-Myc, VEGF, Cyclin D1, and Bcl-2 expressions [83]. In addition, the same researchers postulated a novel mechanism wherein nicotine may promote cell proliferation in HPV16-positive human cervical epithelium cells. In nicotine-treated cells, they observed that the reduction of small ribosomal protein RPS27 expression induced the phosphorylation of ubiquitin-protein ligase Mdm2 and consequently lead to p53 reduction [84]. Moreover, it has been documented that HPV E6 oncoprotein enhances the acknowledged Wnt/β-catenin pathway by inducing FOXM expression through the MZF1/NKX2-1 axis and, consequently, it overexpresses target genes such as Cyclin D and c-Myc. These results may explain a new mechanism whereby HPV may promote invasiveness and stemness in oral and lung cancer cells [85]. Curiously, these findings differ from a recent study in HPV-positive oral carcinoma cells from smoking patients, in which the authors found that the Wnt/βCatenin pathway and target genes (Cyclin D1, Cdh1, Cdkn2a, Cd44, Axin2, c-Myc, and Tcf1) are downregulated in HPV-positive oral cells compared to those HPV-negative. These unexpected results suggest a particular pathological form in HPV-positive oral cancers associated with TS [86]. However, as this was a comparative gene expression study in two oral cell lines, HPV-mediated Wnt/βCatenin activation cannot be denied.
Gene expression alterations in HPV-driven tumorigenesis have been reported which may also be altered by TS. A compelling example of this is the PIR gene, which encodes the redox sensor Pirin and is upregulated by TS in bronchial cells [87]. Likewise, it was observed that PIR is upregulated by HR-HPV E7 oncoprotein dependent on EGFR/MEK/ERK and PI3K/Akt signaling pathways. Subsequently, Pirin protein induces NF-κB activation and is involved in EMT and migration in HPV-positive cervical cancer cells [88]. Taken together, these data indicate that PIR may be important in both TS and HPV-mediated carcinogenesis. A similar role may apply to the redox-sensitive transcription factor NRF2, which is induced by TS and also upregulated in HPV16 positive cervical cancer cells, suggesting it may act as a potential oncogene [89]. Moreover, CXCL14 is a chemotactic factor for dendritic cells, NK cells, and an angiogenesis inhibitor, and it is downregulated in HPV-positive cervical cells and tissues in an E7-dependent manner. Furthermore, oncoprotein E7 triggers CXCL14 promoter hypermethylation. Therefore, these findings suggest a new HPV-mediated oncogenesis route by suppressing antitumor CXCL14 effects [90]. Surprisingly, CXCL14 expression is also highly downregulated in human bronchial epithelial cells exposed to CSE [91]. Altogether, this indicates a common tentative oncogenesis-related factor. In the same way, CXCL12/CXCR4 pathway induces HPV-positive keratinocytes transformation due to high levels of oncoprotein-induced signaling pathways and cell proliferation correlation [92]. Nicotine also stimulates this mechanism by overexpressing CXCR4, thus playing a key role in tumor progression [93]. PTPN14, another candidate gene implicated in metastasis inhibition and negative oncogenesis regulation, has been recently reported as an important mediator in HPV-carcinogenesis. These observations show that HPV16 E7 degrades PTPN14 to inhibit keratinocyte differentiation independent of RB1 inactivation [94]. Another study detected hypophosphorylation of PTPN14 in the presence of TS [95], demonstrating a potential function in HPV-positive cancers. Altogether, a plethora of genes have been shown to be altered in the presence of both TS and HPV, suggesting the possibility of cooperation.
Table 1. Tobacco smoke promotes viral and host-related changes involved in epithelial carcinogenesis1.
|
Molecule |
Effect |
Cell Line |
Ref. |
|
Tobacco Smoke Affects HPV Replication |
|
||
|
BaP |
Increases number of virions and genomes of HPV31 |
CIN (CIN-612 9E) |
[24] |
|
BaP |
Increases number of virions of HPV31 by MAPK ERK1/2 pathway |
CIN (CIN-612 9E) |
[25] |
|
Tobacco Smoke Promotes HPV E6/E7 Expression |
|||
|
Nicotine |
Promotes indirectly HPV16 E6 and E7 expression |
CIN1, CIN2, CIN3 |
[29] |
|
|
|
and cervical cancer |
|
|
BaP |
Increases HPV16 E7 expression |
Cervical carcinoma (CaSki) |
[30] |
|
CSE |
Increases E6 and E7 expression in cells maintaining episomal HPV16 genomes |
CIN1 |
[31] |
|
|
|
(W12 and CIN612) |
|
|
CSE |
Increases tumor properties in cells expressing HPV16 E6 and E7 |
Lung adenocarcinoma (A549) |
[75] |
|
|
|
Lung carcinoma (A549), |
|
|
CSE |
Induces HPV16 p97 promoter activation |
bronquial carcinoma (H-2170), |
[32] |
|
|
|
bronquial (BEAS-2B), alveolar (NL-20), |
|
|
|
|
cervical carcinoma (CaSki and SiHa) |
|
|
CSE |
Promotes mutation that favors AP-1 binding to HPV16 LCR, increasing the activity of p97 promoter |
Oral keratinocyte |
[36] |
|
|
|
(16BNNK) |
|
|
CSE |
Increases HPV16 E6 and E7 through p97 promoter activation involving EGFR/PI3K/Akt/C-Jun signaling pathway |
Cervical carcinoma |
[33] |
|
|
|
(CaSki and SiHa) |
|
|
Tobacco Smoke Promotes DNA Damage |
|
||
|
Nicotine |
Generates adducts and actives Ras signaling pathway |
Lung epithelial (LA4) |
[63] |
|
Acrolein |
Remodels chromatin leading to oxidative stress responses |
Bronchial epithelial (BEAS-2B), |
[76] |
|
|
|
Lung adenocarcinoma (A549) |
|
|
CSE |
Increases DNA damage in cells maintaining episomal HPV16 genomes |
CIN1 |
[31] |
|
|
|
(W12 and CIN612) |
|
|
Tobacco Smoke Affects Immune Responses |
|
||
|
Nicotine |
Induces T cell anergy |
T cells |
[58] |
|
Nicotine |
Promotes migration in HPV18 cells by activating |
Cervical carcinoma |
[60] |
|
|
the PI3K/Akt/NF-κB pathway |
(HeLa) |
|
1Abbreviations: CSE: cigarette smoke extracts; CIN: cervical intraepithelial neoplasia; BaP: benzo[α]pyrene
3. Conclusions and Remarks
HR-HPV infection is a necessary although not sufficient condition for cervical carcinogenesis. Women who smoke are more susceptible to this disease, suggesting that TS is a cofactor. Additionally, although HPV-driven HNCs are a clinical entity distinct from those HPV-negative cases, a portion of HPV-driven HNCs are from subjects who smoke.
Diverse mechanisms have been suggested for a collaboration between HPV and TS in epithelial cancers. First, both TS and HR-HPV affect a plethora of signaling pathways involved in cancer initiation, promotion, and progression, suggesting a complex network of interactions. In addition, experimental approaches demonstrated that TS exposure results in increased E6 and E7 expression and DNA damage in epithelial cells. Moreover, TS affects both innate and adaptative immune responses against HPV. Additionally, specific compounds such as BaP increase HPV replication in cervical cancer cells, with acrolein or nicotine showing immunosuppressive properties. We cannot deny the possibility that additional mechanisms that remain to be discovered may be involved in TS/HPV interactions in cervical and HNCs.
Due to the fact that better survival and outcomes in HPV-positive HNCs has been established, it will be of interest to assess the clinical consequences of TS/HPV interaction in patients with HPV-driven tumors. In addition, other environmental factors or pathogens may be involved in collaboration with HR-HPV for epithelial carcinogenesis. The knowledge of such factors and mechanisms will be important to establish both prevention strategies and to find new therapeutic targets for cervical or HNC treatment. Finally, oncogenic viruses such as HPV are good models to study interactions with environmental or host-related factors for human carcinogenesis.
References
- Bosch, F.X.; De Sanjose, S. The Epidemiology of Human Papillomavirus Infection and Cervical Cancer. Dis. Markers 2007, 23, 213–227, doi:10.1155/2007/914823.
- Wentz, W.B. Histologic grade and survival in cervical cancer. Obstet. Gynecol. 1961, 18, 412–416.
- Arbyn, M.; Weiderpass, E.; Bruni, L.; De Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203, doi:10.1016/s2214-109x(19)30482-6.
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, N.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, doi:10.1002/ijc.29210.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018, 144, 1941–1953, doi:10.1002/ijc.31937.
- De Cecco, L.; Nicolau, M.; Giannoccaro, M.; Daidone, M.G.; Bossi, P.; Locati, L.D.; Licitra, L.; Canevari, S. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget 2015, 6, 9627–9642, doi:10.18632/oncotarget.3301.
- Maasland, D.H.; Brandt, P.A.V.D.; Kremer, B.; Goldbohm, R.A.; Schouten, L.J. Alcohol consumption, cigarette smoking and the risk of subtypes of head-neck cancer: Results from the Netherlands Cohort Study. BMC Cancer 2014, 14, 187, doi:10.1186/1471-2407-14-187.
- Sabatini, M.E.; Chiocca, S. Human papillomavirus as a driver of head and neck cancers. Br. J. Cancer 2019, 122, 306–314, doi:10.1038/s41416-019-0602-7.
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475, doi:10.1158/1055-9965.epi-04-0551.
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus–positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242, doi:10.1200/jco.2015.61.6995.
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35, doi:10.1056/nejmoa0912217.
- Villiers, E.M.; Wagner, D.; Schneider, A.; Wesch, H.; Miklaw, H.; Wahrendorf, J.; Papendick, U.; Hausen, H. Human papillomavirus infections in women with and without abnormal cervical cytology. Lancet 1987, 330, 703–706, doi:10.1016/s0140-6736(87)91072-5.
- Rous, P.; Beard, J.W. A virus-induced mammalian growth with the characters of a tumor (the shope rabbit papilloma). J. Exp. Med. 1934, 60, 741–766, doi:10.1084/jem.60.6.741.
- Winkelstein, W. Smoking and cancer of the uterine cerVIX: Hypothesis. Am. J. Epidemiology 1977, 106, 257–259, doi:10.1093/oxfordjournals.aje.a112460.
- Kim, M.S.; Shin, K.H.; Baek, J.H.; Cherrick, H.M.; Park, N.H. HPV-16, tobacco-specific N-nitrosamine, and N-methyl-N’-nitro-N-nitrosoguanidine in oral carcinogenesis. Cancer Res. 1993, 53, 4811–4816.
- Gunnell, A.S.; Tran, T.N.; Torrång, A.; Dickman, P.W.; Sparén, P.; Palmgren, J.; Ylitalo, N. synergy between cigarette smoking and human papillomavirus type 16 in cervical cancer In situ Development. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2141–2147, doi:10.1158/1055-9965.epi-06-0399.
- Al-Zalabani, A.H. Cancer incidence attributable to tobacco smoking in GCC countries in 2018. Tob. Induc. Dis. 2020, 18, 18, doi:10.18332/tid/118722.
- Louie, K.S.; Castellsagué, X.; De Sanjose, S.; Herrero, R.; Meijer, C.J.; Shah, K.; Muñoz, N.; Bosch, F. For the international agency for research on cancer multicenter cervical cancer study group smoking and passive smoking in cervical cancer risk: Pooled analysis of couples from the IARC multicentric case-control studies. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1379–1390, doi:10.1158/1055-9965.epi-11-0284.
- Plummer, M.; Herrero, R.; Franceschi, S.; Meijer, C.J.; Snijders, P.; Bosch, F.; De Sanjosé, S.; Muñoz, N.; IARC multi-centre cervical cancer study group smoking and cervical cancer: Pooled analysis of the IARC multi-centric case–control study. Cancer Causes Control. 2003, 14, 805–814, doi:10.1023/b:caco.0000003811.98261.3e.
- IARC. Tobacco Smoking and Tobacco Smoke. IARC Monographs on the Evaluation of the Carcinogenic Risks of Chemicals to Humans; IARCPress: Lyon, France, 2004.
- Hang, B. Formation and repair of tobacco carcinogen-derived bulky DNA adducts. J. Nucleic Acids 2010, 2010, 1–29, doi:10.4061/2010/709521.
- A.; Melikian, A.; Sun, P.; Prokopczyk, B.; El-Bayoumy, K.; Hoffmann, D.; Wang, X.; Waggoner, S. Identification of benzo[a]pyrene metabolites in cervical mucus and DNA adducts in cervical tissues in humans by gas chromatography-mass spectrometry. Cancer Lett. 1999, 146, 127–134, doi:10.1016/s0304-3835(99)00203-7.
- Prokopczyk, B.; Cox, J.E.; Hoffmann, D.; E., S.E.S. Identification of tobacco specific carcinogen in the cervical mucus of smokers and nonsmokers. J. Natl. Cancer Inst. 1997, 89, 868–873, doi:10.1093/jnci/89.12.868.
- Alam, S.; Conway, M.J.; Chen, H.-S.; Meyers, C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J. Virol. 2007, 82, 1053–1058, doi:10.1128/jvi.01813-07.
- Bowser, B.S.; Alam, S.; Meyers, C. Treatment of a human papillomavirus type 31b-positive cell line with benzo[a]pyrene increases viral titer through activation of the Erk1/2 signaling pathway. J. Virol. 2011, 85, 4982–4992, doi:10.1128/jvi.00133-11.
- Schiffman, M.; Adrianza, E. ALTS group ASCUS-LSIL triage study. Acta Cytol. 2000, 44, 726–742, doi:10.1159/000328554.
- Xi, L.F.; Koutsky, L.A.; Castle, P.E.; Edelstein, Z.R.; Meyers, C.; Ho, J.; Schiffman, M. Relationship between cigarette smoking and human papilloma virus types 16 and 18 DNA load. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3490–3496, doi:10.1158/1055-9965.EPI-09-0763.
- Castle, E.P.; Meyers, C.; Alam, S.; Conway, M.J. How Does tobacco smoke contribute to cervical carcinogenesis? J. Virol. 2008, 82, 6084–6086, doi:10.1128/jvi.00103-08.
- Ndisang, D.; Khan, A.; Lorenzato, F.; Sindos, M.; Singer, A.; Latchman, D.S. The cellular transcription factor Brn-3a and the smoking-related substance nicotine interact to regulate the activity of the HPV URR in the cervix. Oncogene 2010, 29, 2701–2711, doi:10.1038/onc.2010.33.
- Maher, D.M.; Bell, M.C.; O’Donnell, E.A.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Curcumin suppresses human papillomavirus oncoproteins, restores p53, rb, and ptpn13 proteins and inhibits benzo[a]pyrene-induced upregulation of HPV E7. Mol. Carcinog. 2010, 50, 47–57, doi:10.1002/mc.20695.
- Wei, L.; Griego, A.M.; Chu, M.; Ozbun, M.A. Tobacco exposure results in increased E6 and E7 oncogene expression, DNA damage and mutation rates in cells maintaining episomal human papillomavirus 16 genomes. Carcinogenesis 2014, 35, 2373–2381, doi:10.1093/carcin/bgu156.
- Peña, N.; Carrillo, D.; Muñoz, J.P.; Chnaiderman, J.; Urzúa, U.; Leon, O.; Tornesello, M.L.; Corvalan, A.; Rifo, R.S.; Aguayo, F. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS ONE 2015, 10, e0123029, doi:10.1371/journal.pone.0123029.
- Muñoz, J.P.; Carrillo-Beltrán, D.; Aedo-Aguilera, V.; Calaf, G.M.; León, O.; Maldonado, E.; Tapia, J.C.; Boccardo, E.; Ozbun, M.A.; Aguayo, F. Tobacco exposure enhances human papillomavirus 16 oncogene expression via EGFR/PI3K/Akt/c-Jun signaling pathway in cervical cancer Cells. Front. Microbiol. 2018, 9, doi:10.3389/fmicb.2018.03022.
- Li, Y.T.; He, B.; Wang, Y.Z. Exposure to cigarette smoke upregulates AP-1 activity and induces TNF-alpha overexpression in mouse lungs. Inhal. Toxicol. 2009, 21, 641–647, doi:10.1080/08958370802322596.
- Swenson, W.G.; Wuertz, B.R.K.; Ondrey, F.G. Tobacco carcinogen mediated up-regulation of AP-1 dependent pro-angiogenic cytokines in head and neck carcinogenesis. Mol. Carcinog. 2011, 50, 668–679, doi:10.1002/mc.20775.
- Liu, Y.; Li, J.Z.; Yuan, X.H.; Adler-Storthz, K.; Chen, Z. An AP-1 binding site mutation in HPV-16 LCR enhances E6/E7 promoter activity in human oral epithelial Cells. Virus Genes 2002, 24, 29–37, doi:10.1023/a:1014081803232.
- Chen, Z.; A.; Storthz, K.; Shillitoe, E.J. Mutations in the long control region of human papillomavirus DNA in oral cancer cells, and their functional consequences. Cancer Res. 1997, 57, 1614–1619.
- Geng, H.; Zhao, L.; Liang, Z.; Zhang, Z.; Xie, D.; Bi, L.; Wang, Y.; Zhang, T.; Cheng, L.; Yu, D.; et al. Cigarette smoke extract-induced proliferation of normal human urothelial cells via the MAPK/AP-1 pathway. Oncol. Lett. 2016, 13, 469–475, doi:10.3892/ol.2016.5407.
- Zhong, C.Y.; Zhou, Y.M.; Douglas, G.C.; Witschi, H.; Pinkerton, K.E. MAPK/AP-1 signal pathway in tobacco smoke-induced cell proliferation and squamous metaplasia in the lungs of rats. Carcinogenesis 2005, 26, 2187–2195, doi:10.1093/carcin/bgi189.
- Wang, Y.; Geng, H.; Zhao, L.; Zhang, Z.; Xie, D.; Zhang, T.; Min, J.; Yu, D.; Zhong, C. Role of AP-1 in the tobacco smoke-induced urocystic abnormal cell differentiation and epithelial-mesenchymal transition in vivo. Int J. Clin. Exp. Pathol 2017, 10, 8243–8252.
- Rahman, I.; Antonicelli, F.; MacNee, W. Molecular mechanism of the regulation of glutathione synthesis by tumor necrosis factor-α and dexamethasone in human alveolar epithelial cells. J. Biol. Chem. 1999, 274, 5088–5096, doi:10.1074/jbc.274.8.5088.
- Rahman, I.; Smith, C.A.; Antonicelli, F.D.; MacNee, W. Characterisation of γ-glutamylcysteine synthethase-heavy subunit promoter: A critical role for AP-1. FEBS Lett. 1998, 427, 129–133, doi:10.1016/s0014-5793(98)00410-4.
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; MacNee, W.; Rahman, I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 633–642, doi:10.1165/rcmb.2004-0006oc.
- Filosto, S.; Becker, C.R.; Goldkorn, T. Cigarette smoke induces aberrant EGF receptor activation that mediates lung cancer development and resistance to tyrosine kinase inhibitors. Mol. Cancer Ther. 2012, 11, 795–804, doi:10.1158/1535-7163.mct-11-0698.
- Petecchia, L.; Sabatini, F.; Varesio, L.; Camoirano, A.; Usai, C.; Pezzolo, A.; Rossi, G.A. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest 2009, 135, 1502–1512, doi:10.1378/chest.08-1780.
- A.; Mercer, B.; D’Armiento, J.M. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2006, 1, 137–150, doi:10.2147/copd.2006.1.2.137.
- Sekine, T.; Hirata, T.; Ishikawa, S.; Ito, S.; Ishimori, K.; Matsumura, K.; Muraki, K. Regulation of NRF2, AP‐1 and NF‐κB by cigarette smoke exposure in three‐dimensional human bronchial epithelial cells. J. Appl. Toxicol. 2018, 39, 717–725, doi:10.1002/jat.3761.
- Zhang, Q.; Adiseshaiah, P.; Reddy, S.P. Matrix metalloproteinase/epidermal growth factor receptor/mitogen-activated protein kinase signaling regulatefra-1induction by cigarette smoke in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 72–81, doi:10.1165/rcmb.2004-0198oc.
- Liu, M.; Zhou, C.; Zheng, J. Cigarette smoking impairs the response of EGFR-TKIs therapy in lung adenocarcinoma patients by promoting EGFR signaling and epithelial-mesenchymal transition. Am. J. Transl. Res. 2015, 7, 2026–2035.
- Hu, X.; Peng, N.; Qi, F.; Li, J.; Shi, L.; Chen, R. Cigarette smoke upregulates SPRR3 by favoring c-Jun/Fra1 heterodimerization in human bronchial epithelial cells. Future Oncol. 2018, 14, 2599–2613, doi:10.2217/fon-2018-0043.
- Giuliano, A.R.; Sedjo, R.L.; Roe, D.J.; Harri, R.; Baldwi, S.; Papenfuss, M.R.; Abrahamsen, M.; Inserra, P. Clearance of oncogenic human papillomavirus (HPV) infection: Effect of smoking (United States). Cancer Causes Control 2002, 13, 839–846.
- Koshiol, J.; Schroeder, J.; Jamieson, D.J.; Marshall, S.; Duerr, A.; Heilig, C.M.; Shah, K.V.; Klein, R.S.; Cu-Uvin, S.; Schuman, P.; et al. Smoking and time to clearance of human papillomavirus infection in hiv-seropositive and HIV-seronegative women. Am. J. Epidemiol. 2006, 164, 176–183, doi:10.1093/aje/kwj165.
- Qiu, F.; Liang, C.L.; Liu, H.; Zeng, Y.Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 2016, 8, 268–284, doi:10.18632/oncotarget.13613.
- Ferson, M.; Edwards, A.; Lind, A.; Milton, G.W.; Hersey, P. Low natural killer-cell activity and immunoglobulin levels associated with smoking in human subjects. Int. J. Cancer 1979, 23, 603–609, doi:10.1002/ijc.2910230504.
- Holt, P.G.; Keast, D. Environmentally induced changes in immunological function: Acute and chronic effects of inhalation of tobacco smoke and other atmospheric contaminants in man and experimental animals. Bacteriol. Rev. 1977, 41, 205–216, doi:10.1128/mmbr.41.1.205-216.1977.
- Poppe, W.A.; Ide, P.S.; Drijkoningen, M.P.; Lauweryns, J.M.; Van Assche, A. Tobacco Smoking Impairs the Local Immunosurveillance in the Uterine Cervix. Gynecol. Obstet. Investig. 1995, 39, 34–38, doi:10.1159/000292372.
- Szarewski, A.; Maddox, P.; Royston, P.; Jarvis, M.; Anderson, M.; Guillebaud, J.; Cuzick, J. The effect of stopping smoking on cervical Langerhans’ cells and lymphocytes. BJOG: Int. J. Obstet. Gynaecol. 2001, 108, 295–303.
- Geng, Y.; Savage, S.M.; Razani-Boroujerdi, S.; Sopori, M.L. Effects of nicotine on the immune response. II. Chronic nicotine treatment induces T cell anergy. J. Immunol. 1996, 156, 2384–2390.
- Geng, Y.; Savage, S.; Johnson, L.; Seagrave, J.; Sopori, M. Effects of nicotine on the immune response. i. chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicol. Appl. Pharmacol. 1995, 135, 268–278, doi:10.1006/taap.1995.1233.
- Wang, C.; Gu, W.; Zhang, Y.; Ji, Y.; Wen, Y.; Xu, X. Nicotine promotes cervical carcinoma cell line HeLa migration and invasion by activating PI3k/Akt/NF-κB pathway in vitro. Exp. Toxicol. Pathol. 2017, 69, 402–407, doi:10.1016/j.etp.2017.03.006.
- Lee, W.K.; Ramanathan, M.; Spannhake, E.W.; Lane, A.P. The cigarette smoke component acrolein inhibits expression of the innate immune components IL-8 and human Beta-Defensin 2 by Sinonasal Epithelial cells. Am. J. Rhinol. 2007, 21, 658–663, doi:10.2500/ajr.2007.21.3094.
- Fueldner, C.; Kohlschmidt, J.; Riemschneider, S.; Schulze, F.; Zoldan, K.; Esser, C.; Hauschildt, S.; Lehmann, J. Benzo(a)pyrene attenuates the pattern-recognition-receptor induced proinflammatory phenotype of murine macrophages by inducing IL-10 expression in an aryl hydrocarbon receptor-dependent manner. Toxicol. 2018, 409, 80–90, doi:10.1016/j.tox.2018.07.011.
- Chu, M.; Guo, J.; Chen, C.Y. Long-term Exposure to nicotine, via ras pathway, induces cyclin D1 to stimulate G1Cell cycle transition. J. Biol. Chem. 2004, 280, 6369–6379, doi:10.1074/jbc.m408947200.
- Phillips, D.H.; Schoket, B.; Hewer, A.; Grover, P.L. Human DNA adducts due to smoking and other exposures to carcinogens. Prog. Clin. Biol. Res. 1990, 283–292.
- Phillips, D.H.; Schoket, B.; Hewer, A.; Bailey, E.; Kostic, S.; Vincze, I. Influence of cigarette smoking on the levels of DNA adducts in human bronchial epithelium and white blood cells. Int. J. Cancer 1990, 46, 569–575, doi:10.1002/ijc.2910460403.
- Starek, A.; Podolak, I. Carcinogenic effect of tobacco smoke. Roczniki Państwowego Zakładu Higieny 2009, 60, 299–310.
- Trushin, N.; Alam, S.; El-Bayoumy, K.; Krzeminski, J.; Amin, S.G.; Gullett, J.; Meyers, C.; Prokopczyk, B. Comparative metabolism of benzo[a]pyrene by human keratinocytes infected with high-risk human papillomavirus types 16 and 18 as episomal or integrated genomes. J. Carcinog. 2012, 11, 1, doi:10.4103/1477-3163.92309.
- Gupta, R.C.; Moktar, A.; Ravoori, S.; Vadhanam, M.V.; Gairola, C.G. Cigarette smoke-induced DNA damage and repair detected by the comet assay in HPV-transformed cervical cells. Int. J. Oncol. 2009, 35, 1297–1304, doi:10.3892/ijo_00000447.
- Gupta, R.C.; Moktar, A.; Singh, R.; Vadhanam, M.V.; Ravoori, S.; Lillard, J.W.; Gairola, C.G. Cigarette smoke condensate-induced oxidative DNA damage and its removal in human cervical cancer cells. Int. J. Oncol. 2011, 39, 941–947, doi:10.3892/ijo.2011.1106.
- Chen, B.; Simpson, D.A.; Zhou, Y.; Mitra, A.; Mitchell, D.L.; Cordeiro-Stone, M.; Kaufmann, W.K. Human papilloma virus type16 E6 deregulates CHK1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle 2009, 8, 1775–1787, doi:10.4161/cc.8.11.8724.
- Westra, W.H.; Taube, J.M.; Poeta, M.L.; Begum, S.; Sidransky, D.; Koch, W.M. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2008, 14, 366–369, doi:10.1158/1078-0432.ccr-07-1402.
- Busby-Earle, R.M.; Steel, C.M.; Williams, A.R.; Cohen, B.; Bird, C.C. p53 mutations in cervical carcinogenesis–low frequency and lack of correlation with human papillomavirus status. Br. J. Cancer 1994, 69, 732–737, doi:10.1038/bjc.1994.138.
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713, doi:10.1038/nrc2693.
- Jamaly, S.; Khanehkenari, M.R.; Rao, R.; Patil, G.; Thakur, S.; Ramaswamy, P.; Ajaikumar, B.S.; Sahoo, R. Relationship between p53 overexpression, human papillomavirus infection, and lifestyle in Indian patients with head and neck cancers. Tumor Biol. 2012, 33, 543–550, doi:10.1007/s13277-011-0295-x.
- Muñoz, J.P.; González, C.; Parra, B.; Corvalan, A.; Tornesello, M.L.; Eizuru, Y.; Aguayo, F. Functional interaction between human papillomavirus type 16 E6 and E7 oncoproteins and cigarette smoke components in lung epithelial cells. PLoS ONE 2012, 7, e38178, doi:10.1371/journal.pone.0038178.
- Chen, D.; Fang, L.; Li, H.; Tang, M.S.; Jin, C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation*. J. Biol. Chem. 2013, 288, 21678–21687, doi:10.1074/jbc.M113.476630.
- Doorbar, J. Papillomavirus life cycle organization and biomarker selection. Dis. Markers 2007, 23, 297–313, doi:10.1155/2007/613150.
- Alam, S.; Bowser, B.S.; Conway, M.J.; Israr, M.; Ryndock, E.J.; Xi, L.F.; Meyers, C. Downregulation of Cdc2/CDK1 kinase activity induces the synthesis of noninfectious human papillomavirus type 31b virions in organotypic tissues exposed to benzo[a]pyrene. J. Virol. 2010, 84, 4630–4645, doi:10.1128/jvi.02431-09.
- Zhang, W.; Liu, Y.; Zhao, N.; Chen, H.; Qiao, L.; Zhao, W.; Chen, J.J. Role of Cdk1 in the p53-independent abrogation of the postmitotic checkpoint by human papillomavirus E6. J. Virol. 2014, 89, 2553–2562, doi:10.1128/jvi.02269-14.
- Wang, S.; Pang, T.; Gao, M.; Kang, H.; Ding, W.; Sun, X.; Zhao, Y.; Zhu, W.; Tang, X.; Yao, Y.; et al. HPV E6 induces eIF4E transcription to promote the proliferation and migration of cervical cancer. FEBS Lett. 2013, 587, 690–697, doi:10.1016/j.febslet.2013.01.042.
- Pang, T.; Wang, S.; Gao, M.; Kang, H.; Zhao, Y.; Yao, Y.; Hu, X. HPV18 E7 induces the over-transcription of eIF4E gene in cervical cancer. Iran. J. Basic Med. Sci 2015, 18, 684–690.
- Chen, L.; Wang, H. eIF4E is a critical regulator of human papillomavirus (HPV)-immortalized cervical epithelial (H8) cell growth induced by nicotine. Toxicology 2019, 419, 1–10, doi:10.1016/j.tox.2019.02.017.
- Chen, L.; Wang, H. Nicotine promotes human papillomavirus (HPV)-immortalized cervical epithelial cells (H8) proliferation by activating RPS27a-Mdm2-P53 pathway in vitro. Toxicol. Sci. 2018, 167, 408–418, doi:10.1093/toxsci/kfy246.
- Chen, P.M.; Cheng, Y.W.; Wang, Y.C.; Wu, T.C.; Chen, C.Y.; Lee, H. Up-regulation of FOXM1 by E6 oncoprotein through the MZF1/NKX2-1 axis is required for human papillomavirus-associated tumorigenesis. Neoplasia 2014, 16, 961–971, doi:10.1016/j.neo.2014.09.010.
- Lepore, S.; Lettini, G.; Condelli, V.; Sisinni, L.; Piscazzi, A.; Simeon, V.; Zoppoli, P.; Pedicillo, M.C.; Natalicchio, M.I.; Pietrafesa, M.; et al. comparative gene expression profiling of tobacco-associated HPV-positive versus negative oral squamous carcinoma cell lines. Int. J. Med. Sci. 2020, 17, 112–124, doi:10.7150/ijms.35133.
- Gelbman, B.D.; Heguy, A.; O’Connor, T.P.; Zabner, J.; Crystal, R.G. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir. Res. 2007, 8, 10, doi:10.1186/1465-9921-8-10.
- Carrillo-Beltrán, D.; Muñoz, J.P.; Guerrero-Vásquez, N.; Blanco, R.; León, O.; Lino, V.D.S.; Tapia, J.C.; Maldonado, E.; Dubois-Camacho, K.; Hermoso, M.A.; et al. Human papillomavirus 16 E7 promotes EGFR/PI3K/AKT1/NRF2 signaling pathway contributing to PIR/NF-κB activation in oral cancer cells. Cancers 2020, 12, 1904, doi:10.3390/cancers12071904.
- 159. Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259, doi:10.1172/JCI21146.
- Kontostathi, G.; Zoidakis, J.; Makridakis, M.; Lygirou, V.; Mermelekas, G.; Papadopoulos, T.; Vougas, K.; Vlamis-Gardikas, A.; Drakakis, P.; Loutradis, D.; et al. Cervical cancer cell line secretome highlights the roles of transforming growth factor-beta-induced protein ig-h3, peroxiredoxin-2, and nrf2 on cervical carcinogenesis. BioMed Res. Int. 2017, 2017, 4180703–15, doi:10.1155/2017/4180703.
- Cicchini, L.; Westrich, J.A.; Xu, T.; Vermeer, D.W.; Berger, J.N.; Clambey, E.T.; Lee, D.; Song, J.I.; Lambert, P.F.; Greer, R.O.; et al. Suppression of antitumor immune responses by human papillomavirus through epigenetic downregulation of CXCL14. mBio 2016, 7, e00270–16, doi:10.1128/mbio.00270-16.
- Parsanejad, R.; Fields, W.; Steichen, T.J.; Bombick, B.R.; Doolittle, D.J. Distinct regulatory profiles of interleukins and chemokines in response to cigarette smoke condensate in normal human bronchial epithelial (NHBE) cells. J. Interf. Cytokine Res. 2008, 28, 703–712, doi:10.1089/jir.2008.0139.
- Martínez-García, E.; Irigoyen, M.; González-Moreno, Óscar; Corrales, L.; Teijeira, A.; Salvo, E.; Rouzaut, A. Repetitive nicotine exposure leads to a more malignant and metastasis-prone phenotype of SCLC: A molecular insight into the importance of quitting smoking during treatment. Toxicol. Sci. 2010, 116, 467–476, doi:10.1093/toxsci/kfq138.
- Meuris, F.; Carthagena, L.; Jaracz-Ros, A.; Gaudin, F.; Cutolo, P.; Deback, C.; Xue, Y.; Thierry, F.; Doorbar, J.; Bachelerie, F. The CXCL12/CXCR4 signaling pathway: A new susceptibility factor in human papillomavirus pathogenesis. PLOS Pathog. 2016, 12, e1006039, doi:10.1371/journal.ppat.1006039.
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042, doi:10.1073/pnas.1819534116.
- Solanki, H.S.; Advani, J.; Khan, A.A.; Radhakrishnan, A.; Sahasrabuddhe, N.A.; Pinto, S.M.; Chang, X.; Prasad, T.S.K.; Mathur, P.P.; Sidransky, D.; et al. Chronic cigarette smoke mediated global changes in lung mucoepidermoid cells: A phosphoproteomic analysis. OMICS: A J. Integr. Biol. 2017, 21, 474–487, doi:10.1089/omi.2017.0090.


