 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Grazyna Leszczynska | + 2651 word(s) | 2651 | 2020-07-31 07:38:06 | | | |
| 2 | Bruce Ren | -11 word(s) | 2640 | 2020-08-06 03:43:28 | | | | |
| 3 | Bruce Ren | -11 word(s) | 2640 | 2020-08-06 03:48:45 | | |
Video Upload Options
The review summarizes the methods of site-specific incorporation of nucleobase-modified units into RNA oligomers via the post-synthetic strategy including recently discovered native hypermodified functional groups, fluorescent dyes, photoreactive groups, disulfide crosslinks, and nitroxide spin labels.
The chemical synthesis of modified oligoribonucleotides represents a powerful approach to study the structure, stability, and biological activity of RNAs. Selected RNA modifications have been proven to enhance the drug-like properties of RNA oligomers providing the oligonucleotide-based therapeutic agents in the antisense and siRNA technologies. The important sites of RNA modification/functionalization are the nucleobase residues. Standard phosphoramidite RNA chemistry allows the site-specific incorporation of a large number of functional groups to the nucleobase structure if the building blocks are synthetically obtainable and stable under the conditions of oligonucleotide chemistry and work-up. Otherwise, the chemically modified RNAs are produced by post-synthetic oligoribonucleotide functionalization. This review highlights the post-synthetic RNA modification approach as a convenient and valuable method to introduce a wide variety of nucleobase modifications, including recently discovered native hypermodified functional groups, fluorescent dyes, photoreactive groups, disulfide crosslinks, and nitroxide spin labels.
1. Introduction
Recently, a significant interest has been observed in the use of modified oligoribonucleotides in the fields of molecular biology, biochemistry, and medicine [1][2][3]. The representative sites of RNA modifications are the 5′- and 3′-ends of oligoribonucleotides, and the 2′-positions of the ribose, phosphodiester residues, and nucleobase moieties. Notably, among 150 modified nucleosides identified in cellular RNA sequences, more than 95% of functional groups are installed in the nucleobase heterocycles (Figure 1) [4][5]. Improved mass spectrometry approaches and highly sensitive chemical screens in cellular RNA isolation are still extending this list with new base-modifying units, e.g., cyclic N6-threonylcarbamoyladenosines (ct6A, ms2ct6A) [6][7], 2-methylthiomethylenethio-N6-isopentenyl adenosine (msms2i6A) [8], uniquely lipophilic 5-substituted 2-geranylthiouridines (mnm5ges2U, cmnm5ges2U) [9] and 2-methylthiocytidine (ms2C) [10]. RNA modifications affect the structure, stability, and biological activity of RNA biomolecules, particularly the translation process [11].Experiments of the site-specific incorporation of nucleic base modifications have proved to be a particularly useful method for precisely assessing the role of various functional groups in the structure, function, and biosynthesis of RNA molecules [12][13][14][15][16][17].
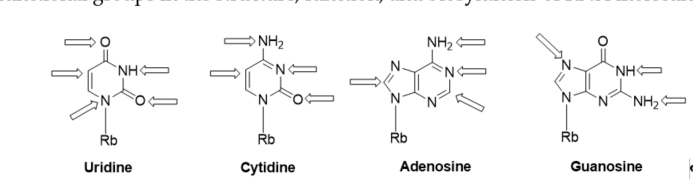
Figure 1. Modified positions distributed in naturally existing RNA nucleobases; Rb: ribose.
Valuable data on RNA structure and cellular activity can be also extracted from biochemical, biophysical, and structural studies involving unnatural base-modified RNA constructs. The insertion of artificial purine/pyrimidine in the place of naturally existing nucleosides may change the local hydrogen bonding system, stacking interactions, or puckering of ribose residues, giving answers as to which modifications or interactions are essential for the functional RNA structure [18][19][20]. Important details on RNA structure, conformational dynamics, and RNA’s homo- and heterotropic interactions can be provided by oligoribonucleotides site-specifically labeled with fluorescent dyes, nitroxide radicals, disulfide crosslinks, or photolabile groups [21][22][23][24][25][26][27][28].
For years, chemically modified oligonucleotides have been extensively studied for the development of new antisense and siRNA therapeutics [29][30][31]. Several nucleobase modifications were found to improve the drug-like properties of oligonucleotides such as cellular uptake, stability in the cell, target specificity, and binding affinity [31][32][33], and reduce undesirable protein binding and immune stimulation [34][35]. Gratifyingly, the incorporation of the first-generation internucleotide linkage modifications and second-generation sugar modifications resulted in the current approval of two RNAi-based therapeutics, Onpattro (patisiran) and Givlaari (givosiran) [31].
Growing interest in the modified RNA oligomers creates the need to develop chemistry that permits the site-specific introduction of functionalizable groups into RNA. The most general way to address the issue of site selectivity is through the use of solid-phase synthesis phosphoramidite chemistry, which involves two strategies: (1) classical modified monomer approach via the preparation of a modified phosphoramidite building block and its subsequent incorporation into the RNA chain; and (2) a post-synthetic RNA modification approach based on selective chemical reaction(s) of precursor unit(s) in the full-length oligonucleotide prepared by the classical method.
This review for the first time summarizes the methods of site-specific incorporation of nucleobase-modified units into RNA oligomers via the post-synthetic strategy. We focused on the nucleobase functionalization due to the high abundance of nucleobase modifications in the nature and the high convenience of the post-synthetic strategy to construct the nucleobase-labeled chemical or biophysical probes. The scope of post-synthetically reactive precursor oligonucleotides has been limited to RNA oligomers prepared by the phosphoramidite chemistry. To facilitate the selection of post-synthetic methods best suited to the assumed chemical–biological objectives, a large number of experimental details have been included.
2. Solid-Phase Synthesis of Modified RNA Oligomers via Phosphoramidite Chemistry
The most current method for the incorporation of a modified nucleoside into a site-specific position of oligoribonucleotides is the phosphoramidite chemistry, which was introduced by Caruthers in 1981 [36][37]. The preparation of RNA oligomers by this strategy involves a four-step reaction cycle including 5′-deblocking, coupling, capping, and oxidation steps (Scheme 1). The synthesis takes the 3′→5′ direction and starts from the 5′-deprotection of fully protected ribonucleoside attached to a solid support (in standard, controlled-pore glass, or polystyrene resins) via the 3′-hydroxyl group. In the second step, activation and coupling, the nucleoside deprived of the 5′-protecting group reacts with a suitably protected phosphoramidite building block activated by the 5-substituted 1H-tetrazole or imidazole derivatives, yielding an unstable phosphite triester linkage. Since a small number of unreacted 5′-hydroxyl sites remain active after coupling, in the third step, capping, the acylating reagent is introduced to prevent them from reacting with the next monomeric unit and elongating the missense strands. In the oxidation step, the support-linked dinucleoside phosphite triester P(III) is converted to the more stable phosphate triester P(V) with an oxidation agent, typically iodine or tert-butylhydroperoxide solutions.
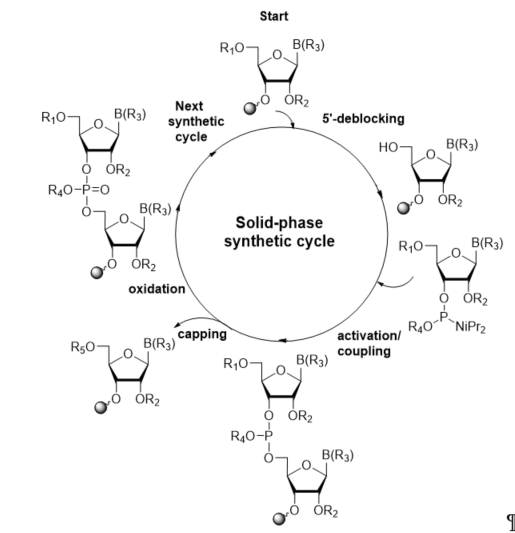
Scheme 1. General scheme of RNA solid-phase synthetic cycle. R1, R2, R4: protecting groups for 5′-OH, 2′-OH, and phosphate residue, respectively; B(R3): protected nucleobase; R5: capping group.
The removal of the 5′-protecting group initiates the next chain extension cycle. Repeating the synthetic cycle provides a full-length oligonucleotide (prepared in the trityl-on or trityl-off mode), which can be released from the solid support and deprotected.
For successful solid-phase RNA synthesis, the phosporamidite monomeric units must be protected with a combination of orthogonal R1 transient and R2, R3, R4 permanent protecting groups. Several synthetic protocols, systems of protecting groups, and deprotection strategies have been demonstrated so far [38]. In practice, the building blocks are protected by employing one of the five most common strategies: (1) 5’-O-DMTr-2’-O-TBDMS [39][40][41]; (2) 5’-O-DMTr-2’-O-TOM [42]; (3) 5’-O-DMTr-2’-O-Fpmp [43]; (4) 5’-O-DMTr-2’-O-TC [44], and (5) 5’-O-DOD(BzH)-2’-O-ACE [45][46] (Figure 2).
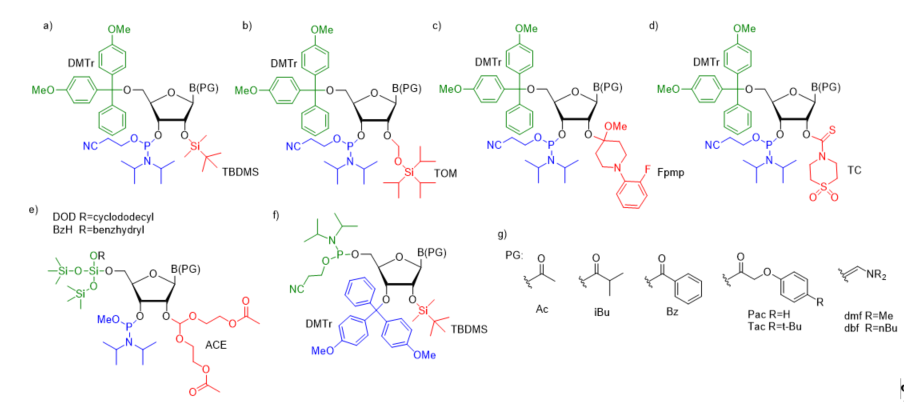
Figure 2. The most common ribonucleoside phosphoramidite building blocks for solid-phase RNA synthesis; (a) 5′-O-DMTr–2′-O-TBDMS–3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite); (b) 5′-O-DMTr–2′-O-TOM–3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite); (c) 5′-O-DMTr–2′-O-Fpmp–3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite); (d) 5′-O-DMTr–2′-O-TC-3′-O-(2-cyano ethyl-N,N-diisopropylphosphoramidite); (e) 5′-O-DOD–2′-O-ACE–3′-O-(methyl-N,N-diisopropyl) phosphoramidite; (f) 3′-O-DMTr-2′-O-TBDMS-5′-O-(2-cyanoethyl-N,N-diisopropylphosphor- amidite) - reverse RNA phosphoramidite; (g) commonly used nucleobase protecting groups.
Most of the synthetic approaches involve 3′-O-β-cyanoethyldiisopropylphosphoramidites containing the transient, acid-labile 5′-O-dimethoxytrityl group (DMTr) and permanent 2′-protection such as silyl blockage (TBDMS or TOM, Figure 2a,b), the acetal masking group (Fpmp, Figure 2c) or carbothioate (TC, Figure 2d). The deprotection of trityl-off RNA oligomers prepared via TBDMS/TOM and Fpmp chemistry starts from the removal of base-labile protecting groups from the phosphotriester backbone and exoamino functions of nucleobases (usually acyl or aminomethylene blockage (Figure 2g). Typically, ammonolytic conditions are used such as aqueous (aq.) ammonia or aq. ammonia–methylamine solution (AMA). This alkaline step of deprotection runs simultaneously with the support cleavage. Optionally, the removal of β-cyanoethyl phosphate groups can be performed separately before the alkaline deprotection step using a weak base in an organic solvent (e.g., TEA/acetonitrile, 1:1 v/v or 10% diethylamine in acetonitrile). Separation of the released acrylonitrile by-product prevents the formation of 2-cyanoethyl-containing oligomer adducts observed when the strong basic conditions of RNA deprotection are used. Removal of the 2′-protecting groups is performed as a final step of oligomer deprotection in the presence of fluoride agent (TBAF or TEA×3HF) for 2′-silyl groups or under acidic conditions (AcOH) when 2′-Fpmp acetal blockage is used. The deprotection of the TC-blocked support-bound oligonucleotide is carried out on a synthesizer column in one step by filling the column with neat ethylenediamine. After TC removal, the “free” oligomer is washed from the column using a small volume of water.
An alternative approach for the orthogonal protection of the 5′- and 2′-hydroxyl groups of monomeric units (prepared as methyl diisopropylphosphoramidites) is based on fluoro-labile 5′-O-silyl protecting groups (DOD or BzH) and acid-labile 2′-O-bis(2-acetoxyethoxy)methyl ortoester protection (ACE, Figure 2e) [45,46]. The exocyclic amino functions of nucleobases are masked with base-labile acyl protecting groups. After 5′-O-DOD/BzH removal (HF/TEA), the oligomer is subjected to a three-step deprotection protocol involving the release of phosphate groups by disodium-2-carbamoyl-2-cyanoethylene-1,1-dithiolate (S2Na2), the removal of base-labile protecting groups, including acetyl groups from 2′-O-ACE blockage using methylamine solution with simultaneous resin cleavage, and finally, the removal of acid-labile 2′-O-bis(2-hydroxyethoxy)methyl orthoesters with AcOH/TEMED.
In addition to the classical phosphoramidite approach proceeding in a 3′→5′ direction, interest has also been focused on the RNA synthesis in the reverse direction (5′→3′) [47][48]. This approach utilizes 5′-O-phosphoramidites (Figure 2f) with a suitable N-protecting group (Bz for adenine, cytosine, Ac for cytosine, and iBu for guanine) and a 3′-O-DMTr-2′-O-TBDMS blockage of sugar residue. Activated 5′-O-phosphoramidite is coupled with a nucleoside bearing a 5′ succinate of ribonucleoside attached to a solid support leading (after oxidation) to the dimer formation with phophodiester linkage and a 3′-O-DMTr-protecting group. The removal of DMTr opens the next synthetic cycle. Oligonucleotide deprotection is performed using the same standard TBDMS chemistry currently utilized in the conventional 3′→5′ synthesis. Although the reverse RNA synthesis is used to a lesser extent than the conventional strategy, it offers a facile route to the assembly of 3′-conjugated RNA constructs of long and medium lengths. Importantly, it was demonstrated that the use of 3′-O-DMTr-5′-phosphoramidites enhances the coupling efficiency, which surpassed 99% per step leading to high-purity RNA products [48]. These distinct advantages of 5′→3′ direction synthesis made the reverse 3′-O-DMTr-5′-phosphoramidites commercially available.
After deprotection, synthetic oligomers (obtained in both conventional and reverse strategies) are purified using polyacrylamide gel electrophoresis (PAGE) or high-performance liquid chromatography (RP HPLC, IE HPLC) and identified by electrospray ionization mass spectrometry (ESI), matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-ToF), and enzymatic digestion analysis.
The solid-phase phosphoramidite method is limited to synthesizing RNA oligomers with lengths of 50–100 nucleotides (longer RNAs molecules can be synthesized by the enzymatic ligation of chemically modified RNA fragment(s) using T4 DNA or T4 RNA ligases [49][50][51] or DNA-zymes [52]). In response to the clinical and commercial success of the therapeutic oligonucleotides, the classical phosphoramidite protocols were translated for use in the large-scale synthesis [53]. Using automated computer-controlled synthesizers, the incorporation of several modifications (phosphorothioate internucleotide linkage, 2′-O-methoxyethyl, 2′-O-Me, 2′-F, locked-, morpholino- and Spiegelmer building blocks) was scaled up from gram- to kilogram-scale production under Current Good Manufacturing Practice (cGMP).
3. Post-Synthetic Strategy for Nucleobase RNA Modifications
First protocols for the post-synthetic functionalization of nucleobases were elaborated for DNA oligomers about three decades ago [54][55][56][57][58][59]. The general concept of post-synthetic RNA modification approach relies on the preparation of an easily convertible/reactive precursor oligonucleotide by the chemical method, e.g., phosphoramidite chemistry, and its subsequent derivatization through chemical reaction(s) involving the reactive center(s) of precursor nucleoside(s). The representative sites of post-synthetic modifications are 5′- and 3′-ends of RNA oligomers, the 2′-position of sugar, phosphate linkage, and nucleobase moieties (Figure 3). The derivatization of nucleobases attends the C-4, C-5, and N-3 positions of uracil, the C-5 position of cytosine, the C-2 position of adenine, and the C-8 and N-7 sites of guanosine (Figure 3). Some of the functional groups have also been attached to the exocyclic amino functions of C, A, and G as well as the thiocarbonyl group of thiouridines (Figure 3).
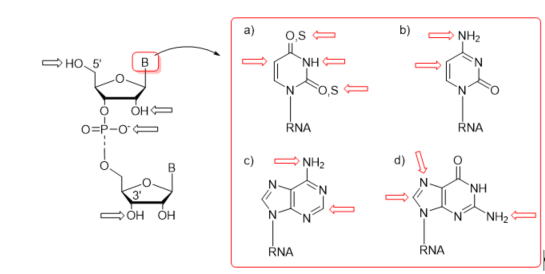
Figure 3. Representative sites of post-synthetic modifications present in sugar–phosphate backbone and nucleobase residues with the highlighting of uridine and thiouridine (a); cytidine (b); adenosine (c); and guanosine (d).
Post-synthetic conversions of RNA oligomers can be performed in the solid or liquid phase (Scheme 2). The “in solid-phase” approach involves a fully protected, support-attached oligoribonucleotide as a substrate for the post-synthetic reaction (Scheme 2A). After conversion, the oligomer is released from the resin and deprotected. Notably, the use of basic conditions for the transformation process can promote the simultaneous removal of exoamino and phosphate-protecting groups as well as the support cleavage. In these cases, the post-synthetic reaction is carried out in a one-step conversion–deprotection process. Occasionally, the separate deprotection of the phosphate groups is performed prior to post-synthetic conversion to avoid any side reactions of the oligomer with acrylonitrile released during removal of the β-cyanoethyl groups.
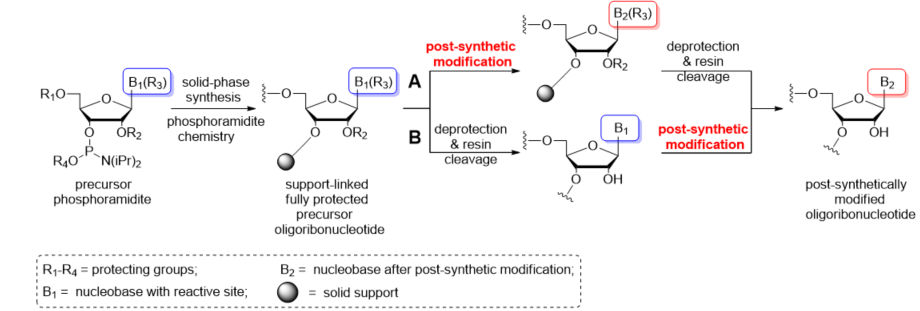
Scheme 2. Schematic representation of the most commonly used post-synthetic protocols of RNA modification: (A) “In solid-phase” approach; (B) “In-solution” approach.
Alternatively, the post-synthetic RNA modification can be carried out in the liquid phase (Scheme 2B). This “in-solution” approach involves a fully deprotected and released precursor oligoribonucleotide. After purification, the “free” oligomer is converted to the target product and repurified to remove the excess of reagents. It is imperative that the presence of free 2′-hydroxyl groups excludes the use of highly alkaline conversion conditions that could promote the strand cleavage or phosphate migration.
Correct incorporation of the final modified unit should be verified by careful analysis of the isolated product by MALDI-ToF, ESI-MS, and/or enzymatic digestion.
The post-synthetic strategy of RNA modification has several important advantages in comparison to the classical modified monomer approach. Firstly, one single modified precursor oligoribonucleotide can provide several sequentially homologous, variously modified RNA fragments [60][61][62]. Such a strategy significantly reduces the cost of synthesis compared to preparing each modified monomeric unit separately. Secondly, hypermodified monomeric units containing highly reactive groups e.g., –COOH, -SO3H, -CHO can be replaced by structurally convenient, easy convertible precursor phosphoramidites deprived of problematic functional groups [62]. Finally, the post-synthetic strategy permits introducing nucleosides/labels that are sensitive to the conditions of solid-phase synthesis and/or oligomer deprotection [63][65]. In some cases, the use of post-synthetic protocol proved to be the method of choice[63][64].
The key to an effective post-synthetic RNA modification strategy is the reactivity of the precursor nucleoside, which should ensure almost quantitative conversion without any perturbation of RNA structure. In addition, the precursor nucleoside should offer an easy approach to afford the phosphoramidite building block and its effective incorporation into the RNA chain. Therefore, the choice of precursor compounds is limited to nucleosides that are fully compatible with the protocols of RNA synthesis and deprotection.
The limited stability and hydrophilic character of oligoribonucleotides significantly reduce the number of organic reactions that can be used in post-synthetic transformations of precursor RNA oligomers. In practice, the conditions of post-synthetic reactions are restricted to polar solvents, moderate temperatures (less than 60 °C), and reaction times shorter than 24 h. Despite the above-mentioned restrictions, the conversions attending the heterocyclic bases represent the highest chemical diversity demonstrated by several types of reactions, including nucleophilic aromatic substitution, allylic substitution, carbon–carbon bond-forming reaction via Sonogashira and Stille couplings, cycloaddition reactions, the derivatization of aliphatic amino groups by the formation of amide bonds, and finally, the functionalization of thiouridines via desulfuration, the formation of thioether bonds, and disulfide bridges. All of the above-mentioned post-synthetic conversions are discussed in the following chapters.
References
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600.
- Röthlisberger, P.; Berk, C.; Hall, J. RNA chemistry for RNA biology. Chimia 2019, 73, 368–373.
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-targeted therapeutics. Cell Metab. 2018, 27, 714–793.
- Machnicka, M.A.; Olchowik, A.; Grosjean, H.; Bujnicki, J.M. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014, 11, 1619–1629.
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; Helm, M.; Bujnicki J.M. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307.
- Matuszewski, M.; Wojciechowski, J.; Miyauchi, K.; Gdaniec, Z.; Wolf, W.M.; Suzuki, T.; Sochacka, E. A hydantoin isoform of cyclic N6-threonylcarbamoyladenosine (ct6A) is present in tRNAs. Nucleic Acids Res. 2017, 45, 2137–2149.
- Kang, B.I.; Miyauchi, K.; Matuszewski, M.; D’Almeida, G.S.; Rubio, M.A.T.; Alfonzo, J.D.; Inoue, K.; Sakaguchi, Y.; Suzuki, T.; Sochacka E.; Suzuki, T. Identification of 2-methylthio cyclic N6 threonylcarbamoyladenosine (ms2ct6A) as a novel RNA modification at position 37 of tRNAs. Nucleic Acids Res. 2017, 45, 2124–2136.
- Dal Magro, C.; Keller, P.; Kotter, A.; Werner, S.; Duarte, V.; Marchand, V.; Ignarski, M.; Freiwald, A.; Müller, R.U.; Dieterich, C.; Motorin, Y.; Butter, F.; Atta, M.; Helm, M. A vastly increased chemical variety of RNA modifications containing a thioacetal structure. Angew. Chem. 2018, 57, 7893–7897.
- Dumelin, C.E.; Chen, Y.; Leconte, A.M.; Chen, Y.G.; Liu, D.R. Discovery and biological characterization of geranylated RNA in bacteria. Nat. Chem. Biol. 2012, 8, 913–919.
- Reichle, V.F.; Petrov, D.P.; Weber, V.; Jung, K.; Kellner, S. NAIL-MS reveals the repair of 2-methylthiocytidine by AlkB in E. coli. Nat. Commun. 2019, 10, doi.org/10.1038/s41467–019–13565–9.
- McCown, P.J.; Ruszkowska, A.; Kunkler, C.N.; Breger, K.; Hulewicz, J.P.; Wang, M.C.; Springer, N.A.; Brown, J.A. Naturally occurring modified ribonucleosides. Wiley Interdiscip. Rev. RNA 2020, e1595.
- Sierant, M.; Leszczynska, G.; Sadowska, K.; Dziergowska, A.; Rozanski, M.; Sochacka, E.; Nawrtot, B. S-Gernayl-2-thiouridine wobble nucleosides of bacterial tRNAs; chemical and enzymatic synthesis of S-geranylated-RNAs and their physicochemical characterization. Nucleic Acids Res. 2016, 44, 10986–10998.
- Sierant, M.; Leszczynska, G.; Sadowska, K.; Komar, P.; Radzikowska-Cieciura, E.; Sochacka, E.; Nawrot; B. Escherichia coli tRNA 2-selenouridine synthase (SelU) converts S2U-RNA to Se2U-RNA via S-geranylated-intermediate. FEBS Lett. 2018, 592, 2248–2258.
- Basanta-Sanchez, M.; Wang, R.; Liu, Z.; Ye, X.; Li, M.; Shi, X.; Agris, P.F.; Zhou, Y.; Huang, Y.; Sheng, J. TET1-mediated oxidation of 5-formylcytosine (5fC) to 5-carboxycytosine (5caC) in RNA. ChemBioChem 2017, 18, 72–76.
- Sun, H.; Sheng, J.; Hassan, A.E.A.; Jiang, S.; Gan, J.; Huang, Z. Novel RNA base pair with higher specificity using single selenium atom. Nucleic Acids Res. 2012, 40, 5171–5179.
- Murphy IV, F.V.; Ramakrishnan, V.; Malkiewicz, A.; Agris, P.F. The role of modifications in codon discrimination by tRNALysUUU. Nat. Struct. Mol. Biol. 2004, 11, 1186–1191.
- Cantara, W.A.; Bilbille, Y.; Kim, J.; Kaiser, R.; Leszczyńska, G.; Malkiewicz, A., Agris, P.F. Modifications modulate anticodon loop dynamics and codon recognition of E. coli tRNA(Arg1,2). J. Mol. Biol. 2012, 416, 579–597.
- Ashraf, S.S.; Ansari, G.; Guenther, R.; Sochacka, E.; Malkiewicz, A.; Agris, P.F. The uridine in “U-turn”: contributions to tRNA-ribosomal binding. RNA 1999, 5, 503–511.
- Erlacher, M.D.; Chirkova, A.; Voegele, P.; Polacek, N. Generation of chemically engineered ribosomes for atomic mutagenesis studies on protein biosynthesis. Nat. Prot. 2011, 6, 580–592.
- Koch, M.; Willi, J.; Pradere, U.; Hall, J.; Polacek, N. Critical 23S rRNA interactions for macrolide-dependent ribosome stalling on the ErmCL nascent peptide chain. Nucleic Acids Res. 2017, 45, 6717–6727.
- Zhang, X.; Cekan, P.; Sigurdsson, S.T.; Qin, P.Z. Studying RNA using site-directed spin-labeling and continuous-wave electron paramagnetic resonance spectroscopy. Methods Enzymol. 2009, 469, 303–328.
- Wojczewski, C.; Stolze, K.; Engels, J.W. Fluorescent oligonucleotides – versatile tools as probes and primers for DNA and RNA analysis. Synlett 1999, 10, 1667–1678.
- Qin, P.Z.; Dieckmann, T. Application of EPR and NMR methods to the study of RNA. Curr. Opin. Struct. Biol. 2004, 14, 350–359.
- Sigurdsson, S.T. Nitroxides and nucleic acids: chemistry and electron paramagnetic resonance (EPR) spectroscopy. Pure Appl. Chem. 2011, 83, 677–686.
- Haller, A.; Soulière, M.F.; Micura, R. The dynamic nature of RNA as key to understanding riboswitch mechanisms. Acc. Chem. Res. 2011, 44, 1339–1348.
- Shelke, S.A.; Sigurdsson, S.T. Site-directed spin labeling of nucleic acids. Eur. J. Org. Chem. 2012, 2012, 2291–2301.
- Wachowius, F.; Höbartner C. Chemical RNA modifications for studies of RNA structure and dynamics. ChemBioChem. 2010, 11, 469–480.
- Hengesbach, M.; Kobitski, A.; Voigts-Hoffmann, F.; Frauer, C.; Nienhaus, G.U.; Helm, M. RNA intramolecular dynamics by single-molecule FRET. Curr. Protoc. Nucleic Acid Chem. 2008, 34, 11.12.1–11.12.22.
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: current progress and future prospects. Chem.Biol. 2012, 19, 60–71.
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644.
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X-J. Therapeutic siRNA: State of the art. Signal Transduct Target Ther. 2020, 5, 101–125.
- Wan, W.B.; Seth, P.P. The medicinal chemistry of therapeutic oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667.
- Watts, J.K.; Deleavey, G.F.; Damha, M.J. Chemically modified siRNA: tools and application. Drug Discov. Today. 2008, 13, 842–855.
- Peacock, H.; Fucini, R.V.; Jayalath, P.; Ibarra-Soza, J.M.; Haringsma, H.J.; Flanagan, W.M.; Willingham, A.; Beal, P.A. Nucleobase and ribose modifications control immunostimulation by a MicroRNA-122-mimetic RNA. J. Am. Chem. Soc. 2011, 133, 9200–9203.
- Peacock, H.; Fostvedt, E.; Beal P.A. Minor-groove-modulating adenosine replacements control protein binding and RNAi activity in siRNA. ACS Chem. Biol. 2010, 5, 1115–1124.
- Beaucage, S.L.; Caruthers, M.H. Deoxynucleoside phosphoramidite. A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22, 1859–1862.
- Matteucci, M.D.; Caruthers, M.H. Synthesis of deoxyoligonucleotides on a polymer support. J. Am. Chem. Soc. 1981, 103, 3185–3191.
- Höbartner, C.; Wachowius, F. Chemical synthesis of modified RNA. In The Chemical Biology of Nucleic Acids; Mayer, B., Ed.; John Wiley & Sons Ltd: Chichester, UK, 2010; pp. 1–37.
- Beaucage, S.L.; Iyer, R.P. The synthesis of modified oligonucleotides by the phosphoramidite approach and their applications. Tetrahedron 1993, 49, 6123–6194.
- Ogilvie, K.K.; Theriault, N.; Sadana, K.L. Synthesis of oligoribonucleotides. J. Am. Chem. Soc. 1977, 99, 7741–7743.
- Ogilvie, K.K.; Beaucagge, S.L.; Schifman, A.L.; Theriault, N.Y.; Sadana, K.L. The synthesis of oligoribonucleotides. II. The use of silyl protecting groups in nucleoside and nucleotide chemistry. VII. Can. J. Chem. 1978, 56, 2768–2780.
- Pitsch, S.; Weiss, P.A.; Jenny, L.; Stutz, A.; Wu, X. Reliable chemical synthesis of oligoribonucleotides (RNA) with 2’-O-[(triisopropylsilyl)oxy]methyl(2’-O-tom)-protected phosphoramidites. Helv. Chim. Acta 2001, 84, 3773–3795.
- Capaldi, D.C.; Reese, C.B. Use of the 1-(2-fluorophenyl)-4-methoxypiperidin-4-yl (Fpmp) and related groups in oligoribonucleotide synthesis: stability of internucleotide linkages to aqueous acid. Nucleic Acids Res. 1994, 22, 2209–2216.
- Dellinger, D.J.; Timar, Z.; Myerson, J.; Sierzchala, A.B.; Turner, J.; Ferreira, F.; Kupihar, Z.; Dellinger, G.; Hill, K.W.; Powell, J.A.; Sampson, J.R.; Caruthers, M.H. Streamlined process for the chemical synthesis of RNA using 2′-O-thionocarbamate-protected nucleoside phosphoramidites in the solid phase. J. Am. Chem. Soc. 2011, 133, 11540–11556.
- Scaringe, S.A.; Wincott, F.E.; Caruthers, M.H. Novel RNA synthesis method using 5′-O-silyl-2′-O-orthoester protecting groups. J. Am. Chem. Soc. 1998, 120, 11820–11821.
- Scaringe, S.A. RNA oligonucleotide synthesis method via 5′-silyl-2′-orthoester chemistry. Methods 2001, 23, 206–217.
- Srivastava, S.C.; Pandey, D.; Srivastava, N.P.; Bajpai, S.P. RNA synthesis: Phosphoramidites for RNA synthesis in the reverse direction. Highly efficient synthesis and application to convenient introduction of ligands, chromophores and modifications of synthetic RNA at the 3′-end. Nucleic Acids Symp. Ser. 2008, 52, 103–104.
- Srivastava, S.C.; Pandey, D.; Srivastava, N.P.; Bajpai, S.P. RNA synthesis by reverse direction process: Phosphoramidites and high purity RNAs and introduction of ligands, chromophores, and modifications at 3′-end. Curr. Protoc. Nucleic Acids Chem. 2011, 45, 3.20.1-3.20.39.
- Duss, O.; Yulikov, M.; Jeschke, G.; Allain, F.H.-T. EPR-aided approach for solution structure determination of large RNAs or protein-RNA complexes. Nat. Commun. 2014, 5, 1–9.
- Kerzhner, M.; Matsuoka, H.; Wuebben, C.; Famulok, M.; Schiemann, O. High yield spin labeling of long RNAs for EPR spectroscopy. Biochemistry 2018, 57, 2923–2931.
- Lang, K.; Micura, R. The preparation of site-specifically modified riboswitch domains as an example for enzymatic ligation of chemically synthesized RNA fragments. Nat. Protoc. 2008, 3, 1457–1466.
- Büttner, L.; Seikowski, J.; Wawrzyniak, K.; Ochmann, A.; Höbartner C. Synthesis of spin-labeled riboswitch RNAa using convertible nucleosides and DNA-catalyzed RNA ligation. Bioorg. Med. Chem. 2013, 21, 6171–6180.
- Sanghvi, Y.S. Large-scale automated synthesis of therapeutic oligonucleotides: a status update. In Advances in Nucleic Acid Therapeutics; Agrawal, S., Gait, M.J. Eds.; Royal Society of Chemistry: London, UK, 2019; Chapter 19, pp. 453–473.
- MacMillan, A.M.; Verdine, G.L. Synthesis of functionally tethered oligodeoxynucleotides by the convertible nucleoside approach. J. Org. Chem. 1990, 55, 5931–5933.
- MacMillan, A.M.; Verdine, G.L. Engineering tethered DNA molecules by the convertible nucleoside approach. Tetrahedron 1991, 47, 2603–2616.
- Harris, C.M.; Zhou, L.; Strand, E.A.; Harris, T.M. New strategy for the synthesis of oligodeoxynucleotides bearing adducts at exocyclic amino sites of purine nucleosides. J. Am. Chem. Soc. 1991, 113, 4328–4329.
- Ferentz, A.E.; Verdine, G.L. Disulfide cross-linked oligonucleotides. J. Am. Chem. Soc. 1991, 113, 4000–4002.
- Ferentz, A.E.; Verdine, G.L. Aminolysis of 2-deoxyinosine aryl ethers: Nucleoside model studies for the synthesis of functionally tethered oligonucleotides. Nucleosides Nucleotides 1992, 11, 1749–1763.
- Xu, Y.Z.; Zheng, Q.; Swann, P.F. Synthesis of DNA containing modified bases by post-synthetic substitution. Synthesis of oligomers containing 4-substituted thymine: O4-alkylthymine, 5-methylcytosine, N4-dimethylamino-5-methylcytosine, and 4-thiothymine. J. Org. Chem. 1992, 57, 3839–3845.
- Kierzek, E.; Kierzek, R. The synthesis of oligonucleotides containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines via post-synthetic modification of precursor oligomers. Nucleic Acids Res. 2003, 31, 4461–4471.
- Chwialkowska, A.; Wielgus, E.; Leszczynska, G.; Sobczak, M.; Mikolajczyk, B.; Sochacka, E.; Nawrot, B. An efficient approach for conversion of 5-substituted 2-thiouridines built in RNA oligomers into corresponding desulfured 4-pyrimidinone products. Bioorg. Med. Chem. Lett. 2015, 25, 3100–3104.
- Bartosik, K.; Sochacka, E.; Leszczynska, G. Post-synthetic conversion of 5-pivaloyloxymethyluridine present in a support-bound RNA oligomer into biologically relevant derivatives of 5-methyluridine. Org. Biomol. Chem. 2017, 15, 2097–2103.
- Matuszewski, M.; Debiec, K.; Sochacka, E. Efficient conversion of N6 threonylcarbamoyladenosine (t6A) into a tRNA native hydantoin cyclic form (ct6A) performed at nucleoside and oligoribonucleotide levels. Chem. Commun. 2017, 53, 7945 7948.
- Debiec, K.; Matuszewski, M.; Podskoczyj, K.; Leszczynska, G.; Sochacka, E. Chemical synthesis of oligoribonucleotide (ASL of tRNALys T. brucei) containing a recently discovered cyclic form of 2-methylthio-N6-threonylcarbamoyladenosine (ct6A). Chem. Eur. J. 2019, 25, 13309–13317.
- Piton, N.; Mu, Y.; Stock, G.; Prisner, T.F.; Schiemann, O.; Engels J.W. Base-specific spin-labeling of RNA for structure determination. Nucleic Acids Res. 2007, 35, 3128–3143.


