 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Walter Alabiso | + 6579 word(s) | 6579 | 2020-07-29 10:34:21 | | | |
| 2 | Walter Alabiso | Meta information modification | 6579 | 2020-07-30 11:30:27 | | | | |
| 3 | Walter Alabiso | Meta information modification | 6579 | 2020-07-30 11:58:56 | | | | |
| 4 | Catherine Yang | -10 word(s) | 6569 | 2020-07-30 12:44:06 | | | | |
| 5 | Catherine Yang | -96 word(s) | 6473 | 2020-10-26 10:48:42 | | |
Video Upload Options
Thermosets are known to be very reliable polymeric materials for high-performance and light-weight applications, due to their retained dimensional stability, chemical inertia and rigidity over a broad range of temperatures. However, once fully cured, they cannot be easily reshaped or reprocessed, thus leaving still unsolved the issues of recycling and the lack of technological flexibility. Vitrimers, introduced by Leibler et al. in 2011, are a valiant step in the direction of bridging the chasm between thermoplastics and thermosets. Owing to their dynamic covalent networks, they can retain mechanical stability and solvent resistance, but can also flow on demand upon heating. More generally, the family of Covalent Adaptable Networks (CANs) is gleaming with astounding potential, thanks to the huge variety of chemistries that may enable bond exchange. Arising from this signature feature, intriguing properties such as self-healing, recyclability and weldability may expand the horizons for thermosets in terms of improved life-span, sustainability and overall enhanced functionality and versatility. In this review, we present a comprehensive overview of the most promising studies featuring CANs and vitrimers specifically, with particular regard for their industrial applications. Investigations into composites and sustainable vitrimers from epoxy-based and elastomeric networks are covered in detail.
1. Introduction
It is common knowledge that polymeric materials break down into two rather broad and diverse major classes: thermoplastics and thermosets. The general features of polymeric materials include chemical stability, thermal and electrical insulation, low cost, light weight, as well as a plethora of tunable properties, which make them appealing for applications in the aerospace, automotive, electronics and biomedical sectors [1][2].
On the one hand, thermoplastics are typically inherently amenable to recycling, reshaping and easy processing due to the non-covalent nature of the bonds holding their chains together. This enables them to flow like a viscoelastic fluid above a specific temperature (or range thereof) and makes them soluble in a good solvent [3][4]. On the other hand, thermosets are insoluble and unable to flow, due to their cross-linked network. Once their shape is set through the so-called curing process, they are further thermally unprocessable and only swell in a good solvent, often with negligible traces of residual dangling chains being eluted away. However, this characteristic is also advantageous, as it imparts attractive properties such as an extended range of thermo-mechanical stability and chemical, wear and creep resistance [5]. Because of their sturdiness, thermosets are vastly deployed for high performance and light-weight applications (e.g., composite materials), though with major concerns regarding non-recyclability and environmental impact [3][4][6][7][8]. As a side note, one may also include elastomers as a thermosetting polymer subclass. These materials are able to withstand a huge deformation thanks to the entropic nature of their elastic recoil [9]. They are very tough and solvent-resistant materials that have been used for industrial applications for decades, such as vulcanized natural rubber for tires [10] or Polydimethylsiloxane (PDMS) for electrical insulation [11]. Over the past decade, many attempts to bridge the gap between thermosets and thermoplastics have been made (see Figure 1), owing to the strong enticement of relevant industrial applications.
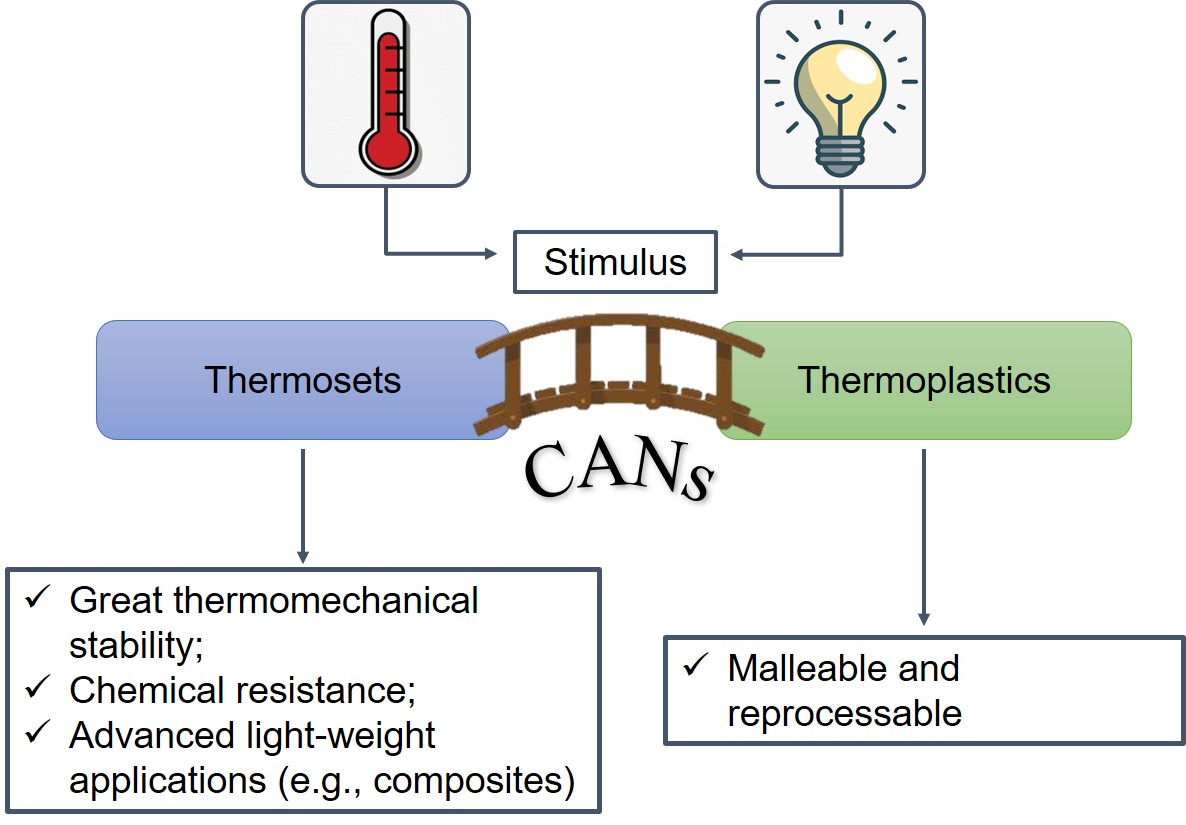
Figure 1. Covalent Adaptable Networks (CANs) as an intermediate bridge between thermosets and thermoplastics.
Getting the best of both worlds would ideally result in a material that retains strong mechanical properties over a large range of temperatures and, at the same time, that is prone to sought-after properties like self-healing, recyclability and weldability.
2. Chemistry of Associative CANs and Vitrimers
As reported in many studies [6][10][12][13][14][15], the most attractive dynamic networks used for vitrimers and vitrimer-like materials rely on exchange reactions like transesterification, transamination of vinylogous acyls (urea/urethanes), transcarbamoylation, transalkylation of triazolium salts, disulfide exchange, imine-amine exchange, siloxane-silanol exchange and olefin metathesis.
2.1. Transesterification
As the name suggests, Transesterification (TE) is a well-known chemical reaction, wherein the organic group of an ester is exchanged with the group of an alcohol (Scheme 1). This reaction is typically catalysed by Brønsted acids [16][17], organo-metallic complexes or organic bases [18].
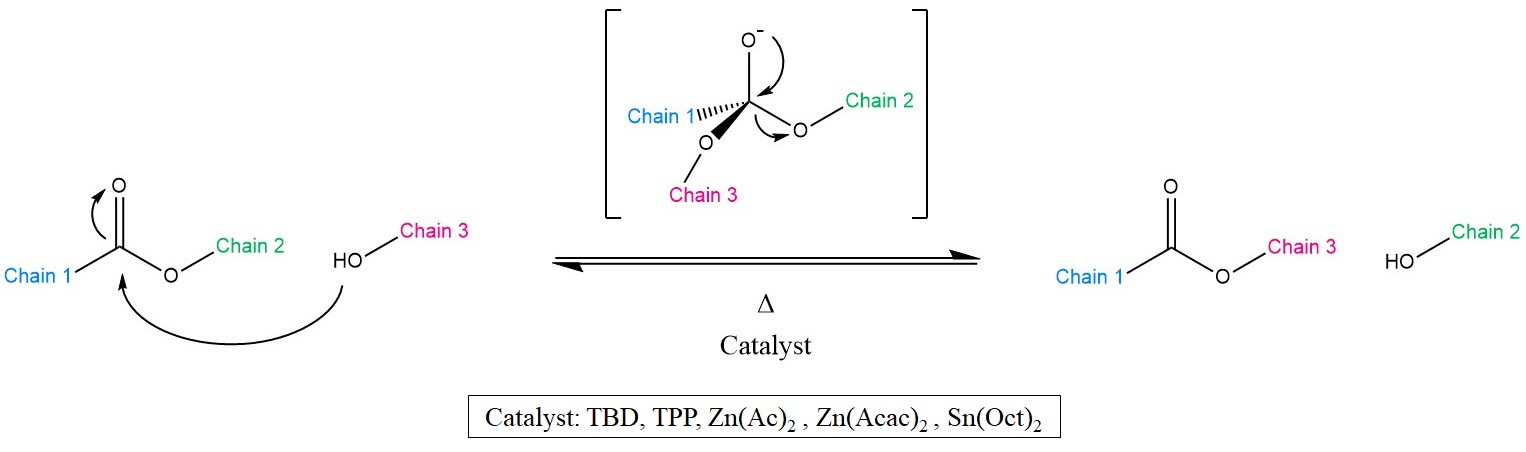
Scheme 1. Representation of the Transesterification (TE) mechanism together with the most common catalysts. As a result, Chains 2 and 3 exchange positions, yet yielding again an alcohol and an ester.
In 2011, Leibler et al. [19] paved the way to vitrimers with a TE-based resin, produced from the classical and well-established chemistry of epoxies. By reacting Diglycidyl Ether of Bisphenol A (DGEBA) with a mixture of fatty tricarboxylic and dicarboxylic acids (epoxy/COOH ratio 1:1) in the presence of Zn(Ac)2, they obtained an insoluble network able to flow and stress-relax at high temperatures. The stress-relaxation took several hours at 150 °C, and the activation energy was estimated to be 80 kJ mol−1 K−1, with a Tv as low as 53 °C for 10 mol% of catalyst. The reprocessability of this network was demonstrated by grinding the material into small bits and by performing injection moulding. Surprisingly, the original mechanical properties were fully restored. Motivated by these first results, in the same paper [19], the authors also tested a formulation based on the epoxy-anhydride system: glutaric anhydride (GA) and DGEBA (1:1 epoxy/acyl ratio) together with 5–10 mol% Zn(Acac)2. Even this system was successfully reprocessed via compression moulding and showed comparable activation energies with respect to the epoxy/acid system.
Since the publication of this study to these days, many other scientists were inspired to propose new ideas and applications for TE. As a matter of fact, epoxy resins based on TE could be very appealing for electronics, adhesives, coatings, light-emitting diodes or as matrices for composites [20]. Further strengthening the applicability of TE-epoxies, many attempts to implement them in advanced materials have been made. Just to mention some: silica-epoxy nanocomposites [14]; high Tg self-healing bio-epoxies from vanillin-guaiacol [21] or eugenol [5]; industrial rubbers and composites thereof [22]. The major strength of transesterification is that it is targeted to commercially-relevant and straight-forward chemicals, connoting a simple implementation. However, the major challenge is to meet the requirements for current thermoplastic processing techniques. The introduction of a simple thermoset resin able to readily flow in a few seconds and undergo several processing cycles would be in fact a revolutionary milestone in polymer engineering.
2.2. Transamination of Vinylogous Acyls
Transamination of vinylogous acyls is a competitive alternative pathway to transesterification. As a matter of fact, the systems based on TE have typically the handicap of not being reprocessable fast enough for the industrial manufacturing standards (e.g., extrusion, injection moulding). The relatively slow stress relaxation leaves compression moulding as the only viable option [23]. Denissen et al. proposed two studies on both vinylogous urethanes [24] and vinylogous urea [25]. The general mechanism is illustrated in Scheme 2.
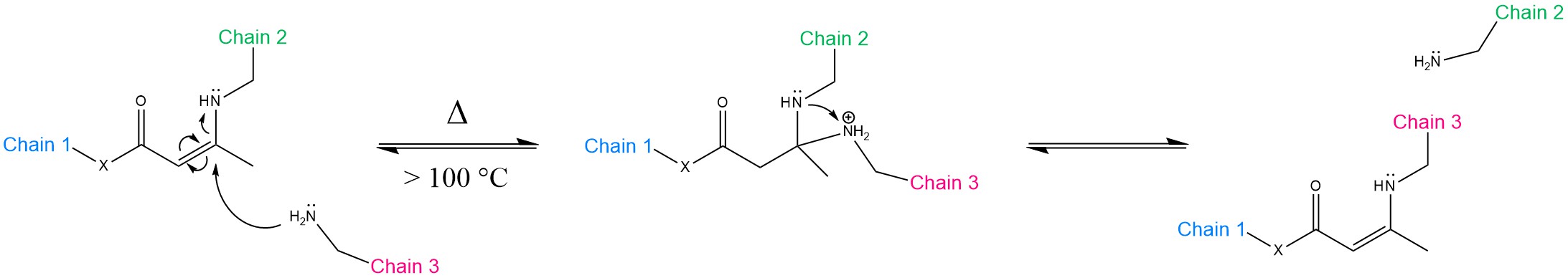
Scheme 2. Mechanism of the transamination of vinylogous acyls. X = CH2, O, NR’R” corresponds to vinylogous amide, urethane and urea, respectively. The reaction can proceed without catalyst at 100 °C or above via the Michael addition pathway. The nucleophilic attack is performed by a free amine group. As a result, Chains 2 and 3 exchange positions.
Because urethanes, ureas and amides are weaker electrophilic groups than esters, regular polyurethane, polyurea and polyamide networks are not prone to readily undergo bond exchange. Instead, the introduction of a vinylic bond inserted between the electron-donating nitrogen and the electron-withdrawing moiety confers Michael-type reactivity and strong resistance to hydrolysis [3][6]. As the name implies, transamination requires free amine groups to proceed, whose stability is crucial to prevent long-term oxidative damage [6]. Nevertheless, the straight-forward synthesis of polyurethanes/polyurea from acetoacetates/acetamide and tertiary amines still represents an attractive path to explore [24][25]. The general trend that Denissen et al. observed in their studies is that the relaxation times decrease substantially as the “X” group becomes more electron-donating (amideurethane →urea) [25]. Whereas vinylogous polyamides were discarded due to their sluggishness, vinylogous polyurethane relaxed in 85 s at 170 °C, and vinylogous polyurea with secondary amide functionality relaxed in only 57 s at the same temperature [25].
Adding a proton donor (a simple acid catalyst such as para-toluenesulfonic acid 0.5–6 mol%) accelerates the exchange reaction even further, therefore meeting the desired industrial standards for thermoplastic manufacturing [25][23]. In fact, vinylogous polyurea matrices were used for the production of glass fibre-reinforced vitrimers via pre-impregnation and thermoforming. The obtained six-ply laminates had a filler content of ca. 57% with E11 of about 40 GPa, comparable to a benchmark epoxy laminate for demanding applications [25]. In another study, Taplan et al. produced an acid-doped vinylogous polyurethane, which, quite remarkably, relaxed in 0.3 s at 160 °C [23]. The material readily lent itself to extrusion, performed at 150 °C with a double-twin screw extruder. The extrudate exhibited a defect-free finish with conserved mechanical and chemical integrity. The slight difference in stress relaxation may be ascribable to partial oxidation of free amine groups. As a final consideration, vinylogous acyls chemistry could definitely offer alternative materials for industry, although they currently suffer from a restricted chemistry and require care in the material preparation.
2.3. Transcarbamoylation
Transcarbamoylation of carbamate groups in polyurethanes may take different pathways, depending on the chemical units involved (Scheme 3). The associative route could occur either via catalysed interaction between two distinct carbamate units or by nucleophilic addition of –OH groups, analogously to transesterification. On the other side, the dissociative route is strongly undesired, as it is often associated with detrimental side-reactions at high temperatures [26]. Because the carbamate group is less nucleophilic than esters, transcarbamoylation is notoriously sluggish [26][27][28][29], with much higher activation energies (Ea = 99–159 kJ mol−1) when compared to transesterification (Ea = 89–163 kJ mol−1) or transamination of vinylogous urethanes (Ea = 31–129 kJ mol−1) [30]. For this reason, getting a sufficient rapidity would necessitate temperatures higher than 200 °C, thereby triggering the adverse reactions mentioned above. Thus, the applicability of transcarbamoylation for reprocessable polyurethanes remains a challenge.
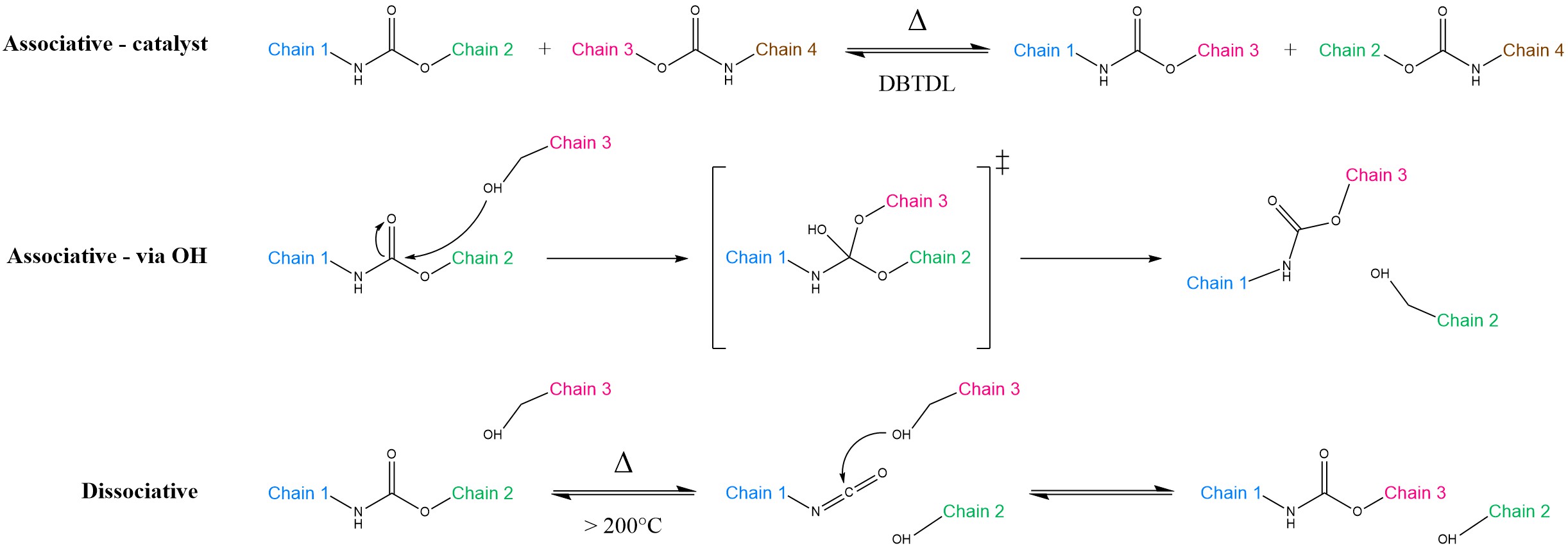
Scheme 3. Transcarbamoylation of urethane units. In the associative-catalyst route (top), Dibutyltin Dilaurate (DBTDL) catalyses the reaction between two carbamate units. In the associative-via OH pathway (middle), a free –OH group performs a nucleophilic attack on the urethane. This is a viable route in polyhydroxyurethanes and resembles transesterification. Instead, in the dissociative mechanism (bottom), the urethane group first splits back into its original isocyanate and alcohol components, with temporary loss of cross-link density. Subsequently, it is able to react again with another –OH group, provided that hydrolysis does not take place first. In all of the cases above, Chains 2 and 3 eventually exchange positions.
Despite this, the doors to intrinsic plasticity in thermoset polyurethanes seem to be still open. Zheng et al. produced a polyurethane based on customary alcohol-diisocyanate chemistry, which is able to undergo comparably fast stress relaxation thanks to an organometallic catalyst (Dibutyltin Dilaurate (DBTDL)) [27]. Notably, the material is able to exhibit both standard elastic shape memory and thermal plasticity. In other words, above the activation temperature for bond exchanges, the network can be rearranged into a new desired permanent shape that is retained upon cooling. Conversely, slightly below the thermal threshold for transcarbamoylation, the thermoset can recover its original shape via the conventional shape memory effect [27]. The same authors improved their work by introducing a tetraol bearing tertiary amines in place of the catalyst [28].
A possible way to overcome the inherent limitations of traditional polyurethanes is to rethink their underlying chemistry. Polyhydroxyurethanes formed by multifunctional cyclic carbonate units and amines unlock the possibility to avoid toxic isocyanate species in the preparation, and they contain free –OH moieties for a supposedly faster bond exchange (Scheme 3, middle) [4][26]. Here, transcarbamoylation may proceed without catalyst and benefits from the concomitance of mechanical and chemical activation [26][31]. On top of that, according to Chen et al., the “non-vitrimeric” coupling of associative transcarbamoylation and dissociative cyclic carbonate aminolysis can definitely boost reprocessability [29].
2.4. Transalkylation
Transalkylation is an interesting mechanism that was employed in the design of ion-conductive vitrimers by Drockenmuller et al. in 2015 [15]. As summarised graphically in Figure 4, a Poly(1,2,3-Triazolium Ionic Liquid) (PTIL) network was synthesised by polyaddition of an α-azide–ω-alkyne monomer via Huisgen 1,3-dipolar Azide–Alkyne Cycloaddition (HAAC) and simultaneous cross-linking with an alkyl halide quaternising agent [15]. As highlighted in Figure 4, bond exchange may occur through two possible pathways. On the one hand, chain reshuffling could be initiated by a nucleophilic attack of the counter-ion on the alkyl-triazolium species (Arrow {1}) with a temporary de-cross-linking [3][6][15]. Such circumstances are reminiscent of a small particle-mediated dissociative pathway [3]. On the other hand, another theorised pathway may involve a concerted SN2-type nucleophilic addition mediated by a free triazole unit (Arrow {2}) [3][6][32].
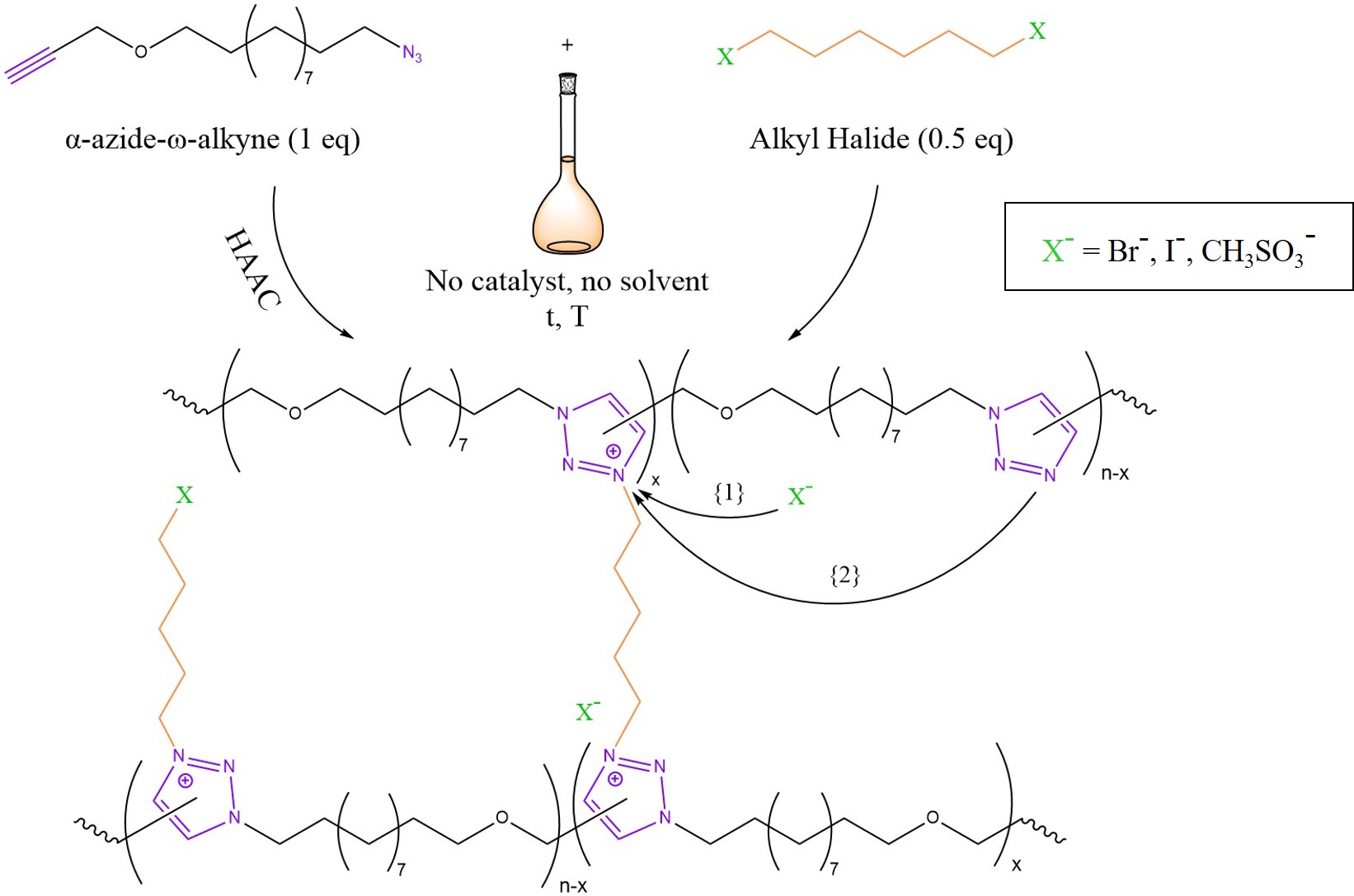
Figure 4. Mechanism of PTILs based on 1,2,3-triazolium salts. The Huisgen 1,3-dipolar Azide–Alkyne Cycloaddition (HAAC) condensation of an α-azide–ω-alkyne monomer takes place in concomitance with cross-linking by means of an alkyl halide. The reaction is one pot and requires no catalyst and no solvent. Possible counter-ions could be bromide (Br−), iodide (I−) or mesylate (CH3SO3−). The two possible bond exchange mechanisms are described in the text. Sketch based on the scientific work of Drockenmuller et al. in [15] and considerations from [3][6].
Therefore, PTILs based on 1,2,3-triazolium salts may be regarded as the missing link between vitrimers and dissociative CANs [30]. The relaxation times of the obtained polymers ranged from 30 min at 130 °C to a few seconds at 200 °C [15]. Most interestingly, this seems to be a consequence of the counter-ion choice where Br− leads to faster relaxation when compared to I− or mesylate [6]. The low viscosity allowed reprocessing via powder compression moulding at 160 °C for 1 h. However, the mechanical properties deteriorated, possibly as a consequence of undesired-side reactions and chain scission. In spite of this limitation, this chemistry seems to be very promising for polyelectrolyte membranes for CO2 recovery and fuel cells or for super-capacitors and batteries as well [15]. Moreover, transalkylation chemistry based instead on sulfonium-sulphide could be promising for sulphur-cured rubbers [33].
2.5. Common Catalysts and the Role Thereof
The choice of a suitable catalyst is undoubtedly linked to the specific system at play. For transesterification, many studies prove the efficiency of organometallic complexes (tin or zinc organic salts) or bases such as Triazabicyclodecene (TBD) and Triphenylphosphine (TPP) [18][34][35]. 1-Methylimidazole was implemented for epoxy-acid systems as well [36]. The main chemicals we are going to cover in this review are illustrated in Figure 5. For vitrimers in general, the main rationale for using a catalyst is to enhance the bond exchange rate, thereby speeding stress relaxation up and tuning the topology freezing transition broadness (see Figure 6a) [34]. Naturally, the downsides would be a more complicated and more costly formulation, as well as the need to guarantee a homogeneous mixture. Several patents regarding vitrimer + catalyst formulations have been published, indicative of the strong potential applicability of vitrimers in the industrial sector [37][37][38].
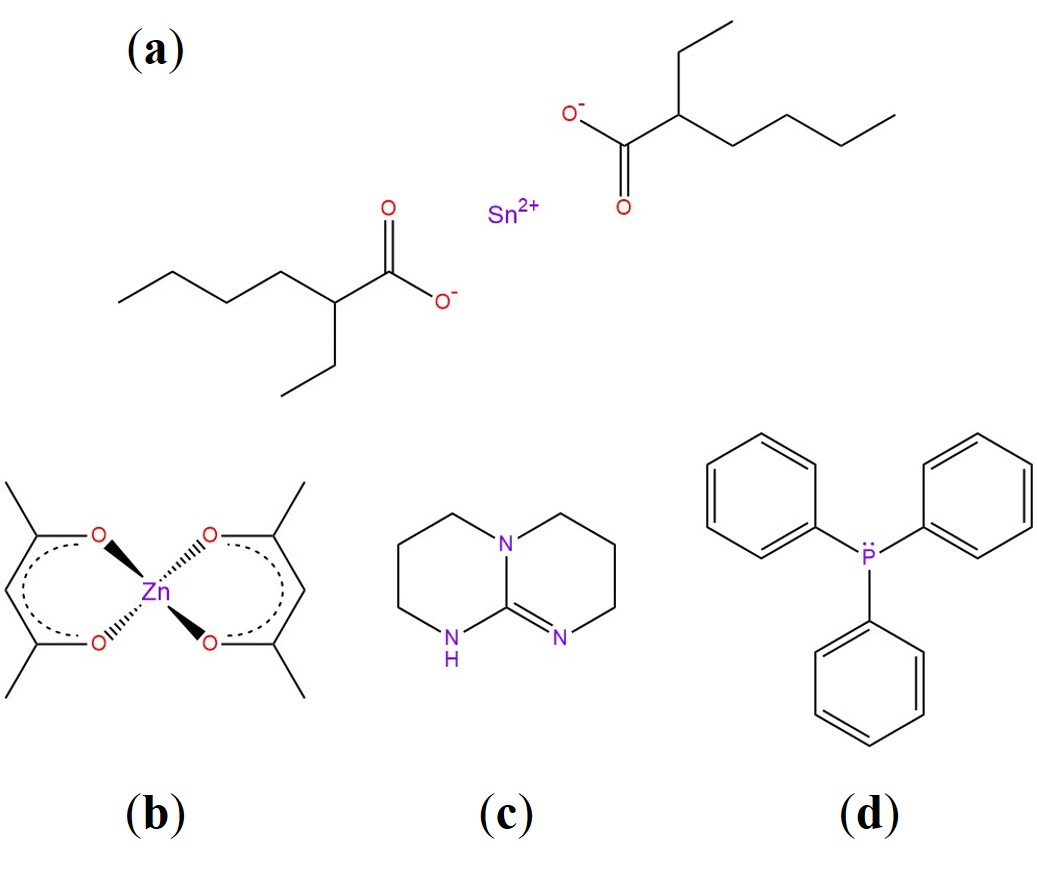
Figure 5. (a) Tin (II) 2-ethylhexanoate, Stannous Octoate (Sn(Oct)2). (b) Zinc acetylacetonate (Zn(Acac)2). (c) Triazabicyclodecene (TBD). (d) Triphenylphosphine (TPP).
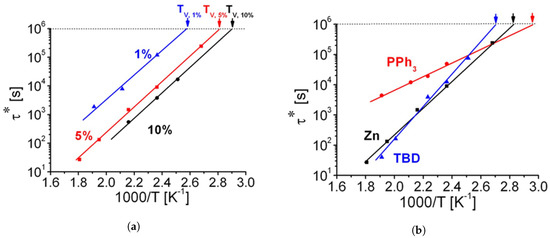
Figure 6. (a) Arrhenius plot of the measured relaxation times for 1, 5, and 10 mol% of Zn(Ac)2. As the concentration increases, Tv shifts to lower temperatures, and the relaxation time is shorter. The activation energy (slope of the curve) is dependent on the system, and therefore, it stays unchanged. (b) Arrhenius plot of the relaxation times for Zn(Ac)2, TPP (labelled as PPh3 by the original authors) and TBD at 5 mol%, measured from shear stress relaxation experiments. It is evident that the system containing TPP has lower Tv, and it shows a lower reactivity, whereas TBD and Zn(Ac)2 have a comparable performance. Adapted with permission from [34]. Copyright 2012©, American Chemical Society, ACS.
Zinc Acetylacetonate (Zn(Acac)2). Thanks to its high thermal resistance, Zn(Acac)2 and similar zinc-based organometallic catalysts became the golden standard for transesterification chemistry, as reported in a plethora of studies [19][39][40][20][14][41]. The main role of zinc catalysts is threefold: (1) bring the reactive species together through directional coordination bonds, (2) increase the electrophilicity of the carbonyl group in esters by polarisation and (3) shift the alcohol/alkoxide equilibrium toward the more nucleophile alkoxide [42]. All these actions underlie β-hydroxy ester formation and transesterification [43]. A clever route was also explored by Niu et al. in a recent paper [44], where they produced a random copolymer of zinc methacrylate and acrylonitrile as a catalyst for DGEBA and sebacic acid. They demonstrated that the catalyst is more efficient than TBD and conventional Zn(Ac)2 under the same loading content (only ~0.75 mol%).
Triazabicyclodecene (TBD). TBD is a strong guanidine base that found application in vitrimer-like elastomers from nitrile-butadiene rubber [22], graphene-vitrimer composites [45] and photo-weldable carbon nanotube-reinforced vitrimeric matrices [46]. TBD is able to enhance the nucleophilicity of the alcoholic group via H-bond, and therein lies its high efficiency as a transesterification catalyst [47].
Triphenylphosphine (TPP). TPP can actively catalyse transesterification due to the lone pair available on its phosphorous atom [48]. When compared to Zn(Acac)2 and TBD, it has a lower activation energy and lower Tv, promoting an overall more sluggish bond exchange rate (Figure 6b) [34]. To the best of our knowledge, the potential of TPP in vitrimers remains mostly unexplored.
Stannous Octoate (Sn(Oct)2). Brutman et al. designed a polylactide vitrimeric network, cross-linked with a diisocyanate in the presence of Sn(Oct)2 [49]. The catalyst is known for its catalytic activity in transesterification, high stability and low toxicity [49]. Crucially, the network rearrangement could occur even in the presence of a low number of free –OH groups. The delicate balance between catalyst load and the free isocyanate:OH ratio could significantly affect the properties of the network.
Dibutyltin Dilaurate (DBTDL). Lastly, dibutyltin dilaurate, has been used for transcarbamoylation. The polarisation of the isocyanate moiety facilitates the reaction with the alcohol [50], which can be useful both for catalysing cross-linking and transcarbamoylation (Scheme 4). A study suggested that DBTDL may also be useful for transesterification of epoxy-anhydride systems [51].
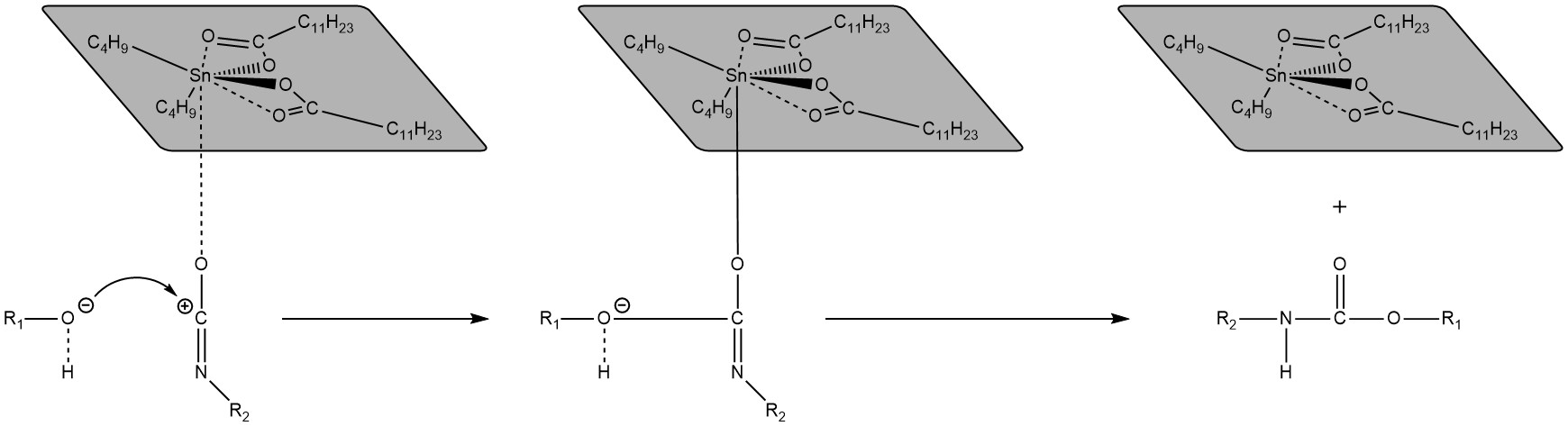
Scheme 4. Illustration of the plausible mechanism of dibutyltin dilaurate as a catalyst for the cross-linking and transcarbamoylation of polyurethanes. Sketch based on considerations from [50].
For alternative chemistries, one may want to deviate from the well-established catalysts found in literature. Snyder et al. reported the synthesis of polycarbonate vitrimers undergoing transcarbonation [52]. Polycarbonate has a great potential for sustainability, and it is massively employed (up to three million tons per year) for resistant and high-impact windows and safety glasses [52]. Transcarbonation occurs, mutatis mutandis, analogously to transesterification, with carbonate units reshuffling upon reaction with free –OH groups. Snyder et al. proved that Titanium (IV) isopropoxide (Ti(Oi-Pr)4) successfully catalyses a vitrimer from 1,4-butanediol and bis-cyclic carbonate (activation energies as low as 81 ± 3 kJ mol−1 and relaxation times as low as τ* = 200 s for 4 mol% of catalyst) [52]. In addition to that, the combination of a Ti(Oi-Pr)4-catalysed polycarbonate vitrimer matrix and natural cellulose has been proven appealing for novel green composites [53]. As a side note, Hatano and coworkers recently endeavoured to introduce a metal-free transesterification catalyst based on tetramethylammonium methyl carbonate [54].
3. State-of-the-Art of Industrially-Relevant Applications
3.1. Examples and Applications of dissociative CANs—Diels–Alder Reaction
In this subsection, we present perhaps the most notable example of dissociative CAN chemistry with a promising scope in the industrial sector: Diels–Alder (DA) chemistry. The prototypical reaction, presented in Scheme 5, involves a [4+2] cycloaddition between an electron-rich diene and an electron-poor dienophile [4][32].
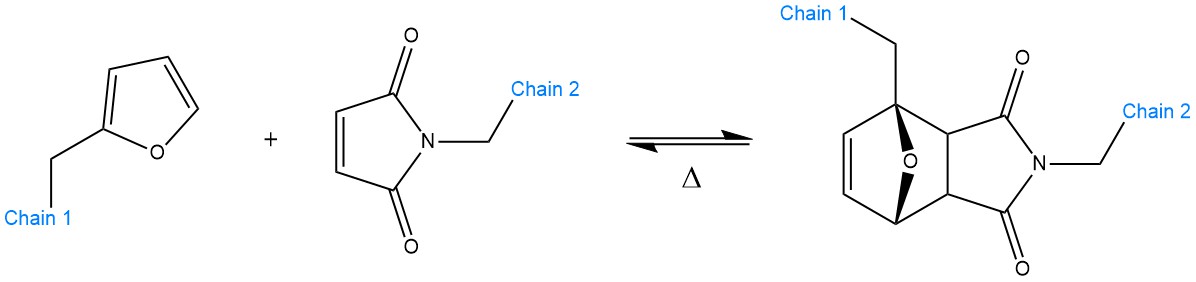
Scheme 5. Schematic representation of a Diels–Alder reaction between a furan (left) and a maleimide (right) unit. When such functional groups are grafted on polymeric chains (Chains 1 and 2), they can enable reversible cross-linking. The reaction can revert back, typically, at 120–130 °C or above. The equilibrium constant Δ depends on the chemistry at play.
At elevated temperatures, the dynamic equilibrium can be shifted back toward the unbound state (retro-DA reaction) in a “click-like” fashion. A classical example of two interacting moieties are furan (diene) and maleimide (dienophile with electron-withdrawing ketone groups). Even though the DA cycloaddition is a fairly well-known process in chemistry (i.e., for cross-linking of polystyrene with furan-maleimide by Stevens and Jenkins in 1979 [4]), it was not until 2002, with the pioneering work of Wudl et al., that it gained popularity [4][32]. The network they obtained from tetrafunctional furan monomers and trifunctional maleimide units proved to be an appealing paradigm for sought-after properties such as shape memory, reprocessability and self-healing [4].
The shape memory effect may be used to confer solid state plasticity on thermosets, whereby one can achieve the manipulation of complex geometry and shape memory multifunctional devices. For instance, Rivero et al. designed a polyurethane network, wherein the shape memory property of polycaprolactone (PCL) units in the backbone assisted self-healing (SMASH). By applying heat, the synergistic effect of reversible furan-maleimide cross-linking units and elastic recoil favours rapid crack closure and network rearrangement Figure 7.
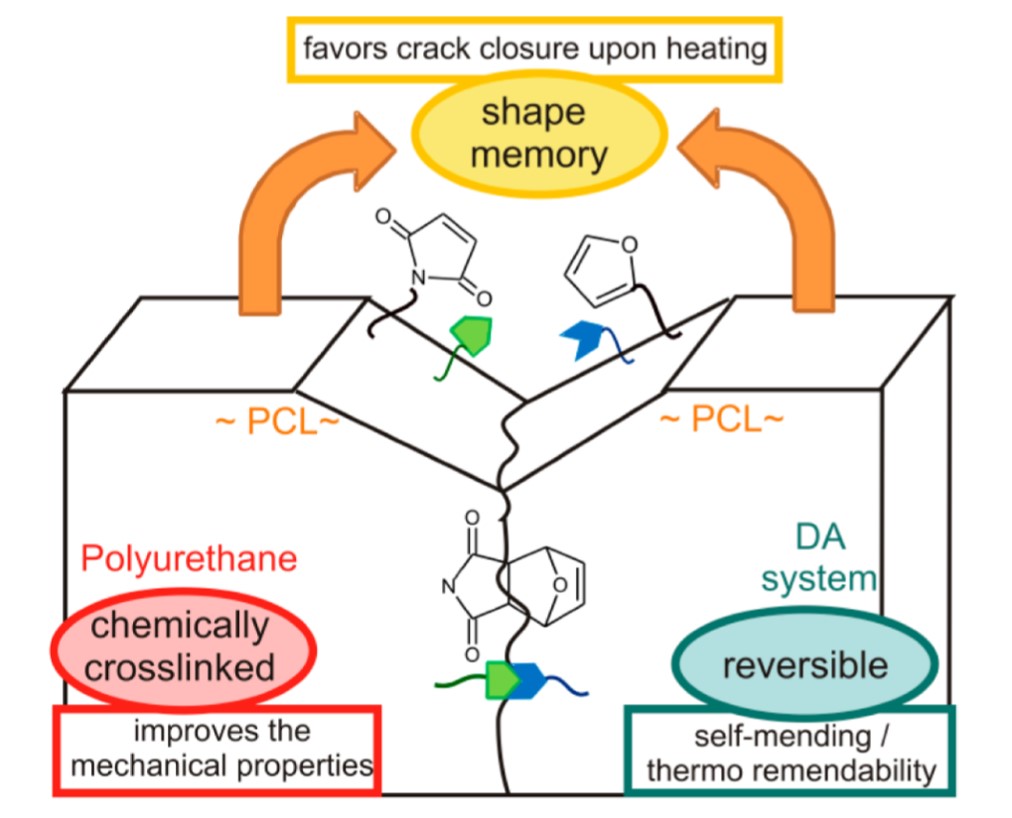
Figure 7. Schematic representation of crack closure as a synergistic effect of the shape memory effect and furan-maleimide chemistry in a PCL-based polyurethane. Reproduced with permission from [55]. Copyright 2014©, American Chemical Society, ACS.
This “self-sewing” effect restores the original mechanical properties: the authors reported excellent healing of macro-scratches after one day at 50 °C [55]. Such behaviour is a function of the furan-maleimide groups density, as well as the content of hard segments (i.e., cross-link density, polycaprolactone segments) of the material. Another promising study presented by Billiet et al. introduced the concept of “click-transclick” chemistry applied to reversible Triazolinedione-indole (TAD-indole) Diels–Alder-like reactions [56]. The principle, presented here in Scheme 6, consists of a first click reaction between a TAD unit and indole, which proceeds spontaneously at room temperature with high yields and no need for a catalyst or any external stimuli (Scheme 6a) [56][57]. The now bound state can undergo an irreversible “transclick” reaction with a diene, thereby yielding the original indole and a new TAD-diene adduct (Scheme 6b). The authors applied this new concept to both a high Tg polymethyl methacrylate and a polyurethane, by means of a difunctional-TAD cross-linker [56].
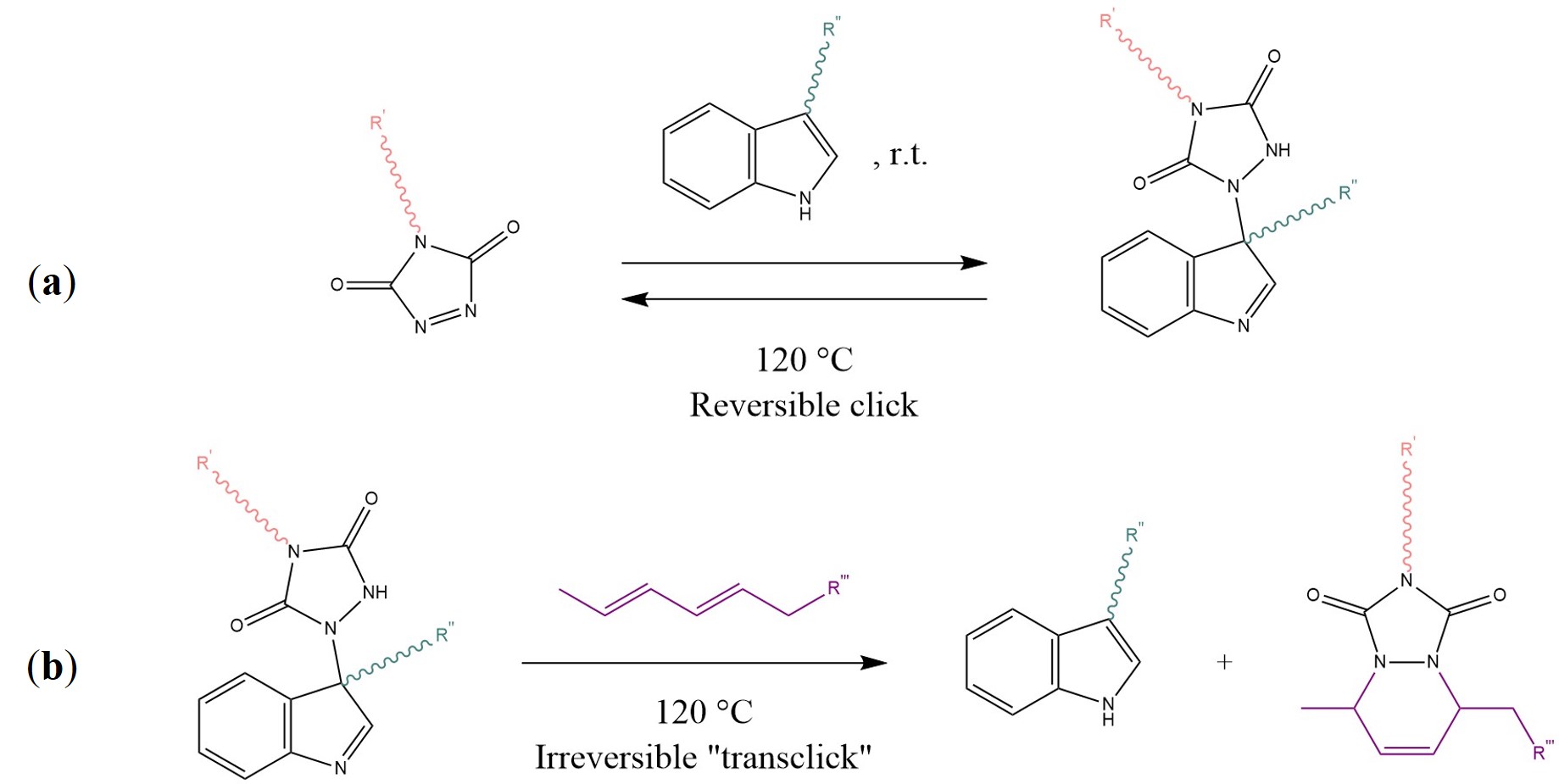
Scheme 6. (a) Reaction between 1,2,4-Triazoline-3,5-Dione (TAD) and an indole at room temperature with a reversible click mechanism. (b) Irreversible transclick reaction between the TAD-indole adduct and a diene, with the formation of the original indole and a TAD-diene adduct. Sketch based on considerations from [56][57].
The two networks could be reprocessed in a mould by extrusion and by compression, respectively. Recently, this knowledge stepped further from niche chemistry to concrete potential in nanostructured polymer substrates, useful in biosensing, information storage, electronics, and anti-fouling coatings [57]. Roling et al. designed some rewritable polymer brush micro-patterns [57], as represented in Figure 8. By preparing an indole-functionalised surface, they were able to graft a TAD-bearing Atom Transfer Radical Polymerisation (ATRP) initiator in a predefined pattern with a PDMS mould (①). By triggering the Surface-Induced reaction (SI-ATRP), they grew polymethacrylate micro-brushes. Subsequently, it was possible to reclaim the original indole surface by means of an erasing procedure via TAD-diene transclick reaction (e.g., with either a 2,4-hexadien1-ol or a -phellandrene solution) (②).
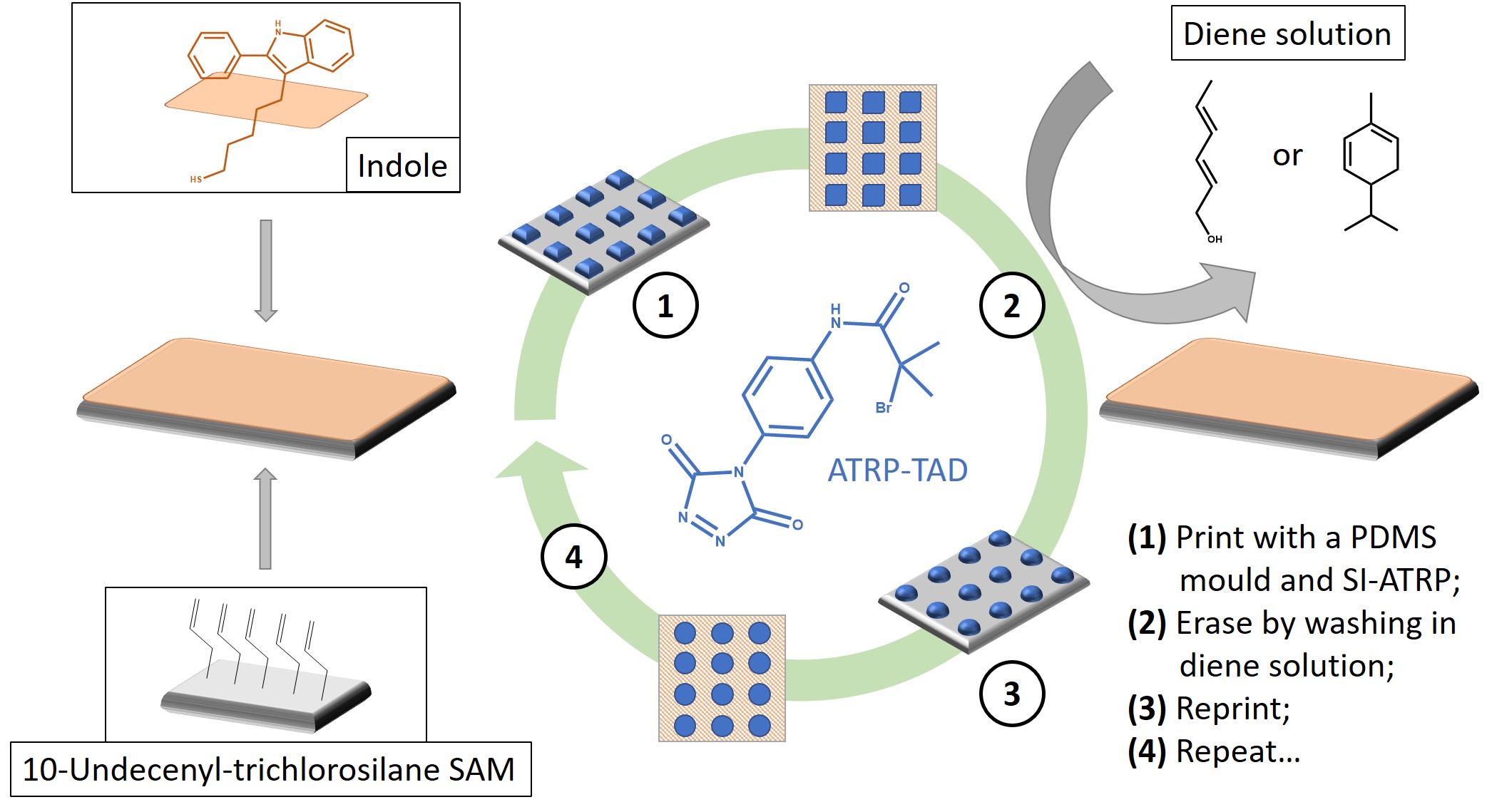
Figure 8. Schematic representation of the rewritable polymer brushes as per the original design of Roling et al. The explanation is available in the picture and in the text. SAM = Self-Assembled Monolayer. Sketch based on considerations from [57]. SI-ATRP, Surface-Induced Atom Transfer Radical Polymerisation.
The quasi-pristine new surface was then amenable to multiple re-printing cycles with excellent spatial control (③ and ④). All of the studies mentioned above are clear examples of engineering materials where the dissociative nature of networks may be highly desirable.
3.2. Applications of Associative CANs and Vitrimers in the Industrial World
3.2.1. Sustainable Vitrimers
With the soaring demand for environmentally-friendly materials and sustainable technology, researchers set out to harness bio-renewable sources. Quite interestingly, some studies demonstrated that vitrimers lend themselves to such purposes.
Lignin is the second most abundant polymer on Earth [58]. Most of the available technical lignin comes from residues of the paper making process [59], and represents a vastly underexploited source of biomass, with over 50 million tons produced per year [60]. Its polyphenolic structure seems to be promising for the formulation of alternative phenol-formaldehyde resins [59][61][62] and bio-epoxy asphalt [63], and it has been utilised in vitrimeric recoverable adhesives [58]. Nevertheless, owing to the low reactivity and low purity of the chemicals contained in lignin waste, some additional thermochemical treatments and costs are required (e.g., methylolation, phenolation) [59]. Therefore, the concrete inclusion of lignin in industry faces some practical hurdles. Zhang et al. prepared an adhesive based on catalyst-assisted transesterification between ozonated lignin and epoxidised sebacic acid [58]. For the highest lignin-to-epoxy ratio, the material showed the highest Tg (133 °C), fast relaxation (τ* = 81 s) and highest strength. More interestingly, the adhesive underwent a cohesive failure at 6.5 MPa when used to bond two aluminium sheets, which is comparable with commercial epoxy adhesives applied to join aluminium alloys [58]. Transesterification helped to achieve multiple bonding cycles with fairly satisfying consistency
It is common knowledge in polymer engineering that Bisphenol A (BPA) has been deemed carcinogenic, toxic and endocrine-disrupting [64]. Therefore, the attention of industries is gradually shifting toward alternative epoxies for thermosets and composites. In the light of this, here we report studies on triglycidyl ether of phloroglucinol extracted from algae, polyimine networks from vanillin [65] or fructose [66] and derivatives of lignin [60]. In particular, diglycidyl ethers of vanillyl alcohol and bisguaiacol show great mobility, stiffness and thermal resistance (comparable to commercial BPA-based resins, such as EPON 828 and EPON 862) when cross-linked with a cycloaliphatic diamine [60].
Cellulose is another abundant natural resource that has also found application in reinforced vitrimers, such as high water resistance nanopaper [35] and vitrimer-paper composites [53]. Zhao et al. focused on the latter application and proposed a transcarbonation-based polycarbonate matrix filled with a cellulose framework [53]. The dynamic nature of the matrix provided facile recyclability, reprocessability and self-healing. Besides, bond exchange in conjunction with hot pressing allowed the production of compact laminates with significantly improved mechanical properties (Figure 9). In addition to that, the authors soaked a criss-cross scratched vitrimer paper sheet in a solution of isopropyl alcohol containing Silver Nanowires (AgNW). The conductive fillers piled up preferentially into the notches. After healing the regions back at 160 °C, they obtained a foil with customised conductive pathways.
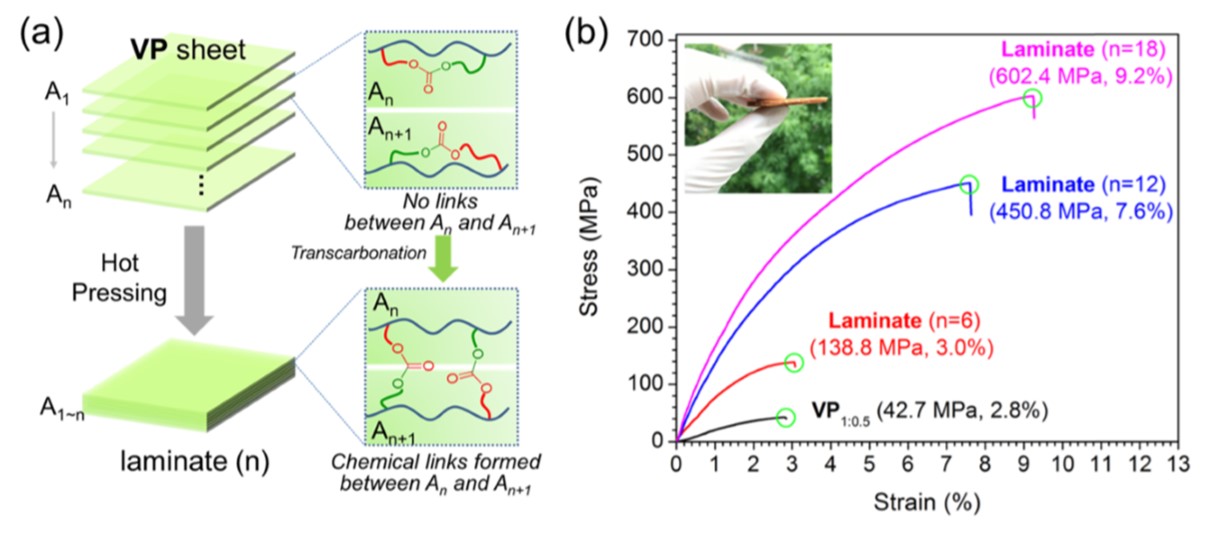
Figure 9. Demonstration of the reprocessing ability of curved Vitrimer-Paper (VP) composites using VP1:0.5 (bis-cyclic carbonate to propanediol ratio) sheets to prepare VP laminates via hot pressing. (a) Diagram of laminate preparation. (b) Tensile stress–strain curves of resultant laminates. Reproduced with permission from [53]. Copyright 2019©, American Chemical Society, ACS.
Another massively underutilised natural resource is crustacean shell waste, available to the extent of six to eight million tons per year [67]. Crab and lobster shells are made out of chitin (15–40%) [67], a polysaccharide that, along with its deacetylated derivative chitosan, has tremendous utility in biotechnology, environmental protection, pharmaceutics and nutrition [68][69]. Ghosh et al. developed a self-healing polyurethane based on an oxetane-substituted derivative of chitosan and a trifunctional isocyanate [70]]. The network was able to self-heal in 30 min upon UV irradiation (280 to 400 nm) thanks to bond reshuffling enabled by the oxetane moiety. Lastly, vitrimer chemistry may be extremely useful for the production of green epoxidised-rubber composites. The release of toxic volatile organic compounds coming from the curing additives used in the rubber industry poses a serious hazard to health, as well as to the environment [41]. Zhang and coworkers synthesised a rubber via in situ epoxidation of an ethylene-propylene-diene monomer [41]. The cross-linking with bio-based decanedioic acid provided a matrix prone to undergo transesterification, which was then reinforced with carbon black. It was possible to recycle the composite via hot pressing a blend of rubber scrap with the uncured formulation. The shape memory effect, facile recycling with no need for grinding and non-toxic curing make this study a promising paradigm for green rubbers.
3.2.2. Epoxy Resins
Epoxy/anhydride systems. Using an anhydride as a hardener for epoxy thermosets is a rather popular industrial choice. The scope of this formulation includes, but is not limited to: LEDs, encapsulation of electronic parts, conductive ink paste and adhesives in structural components produced via RTM, where a high Tg is desirable [71]. The most plausible reaction mechanism (Scheme 7) consists of the alternating ring opening of the anhydride ring and oxirane ring (i.e., epoxide ring), resulting in a stiff polyester [20].
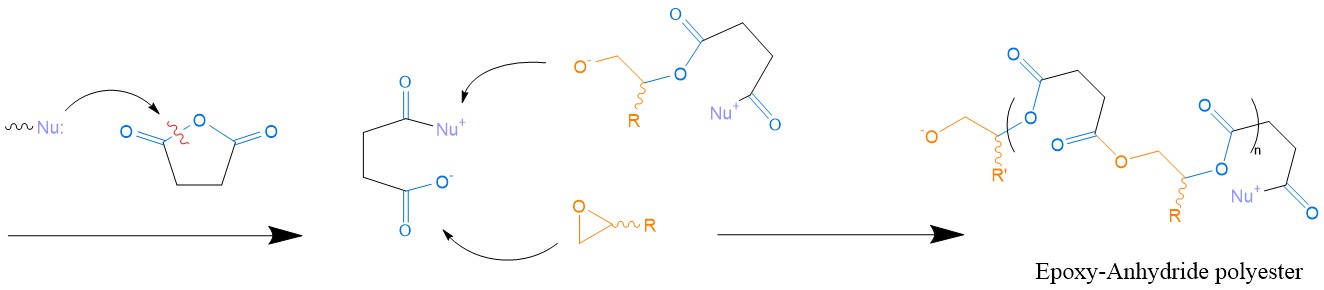
Scheme 7. Mechanism of epoxy/anhydride curing (here with succinic anhydride). The reaction takes place via successive oxirane-anhydride ring opening with or without the aid of a catalyst. The final network could be regarded as a polyester/polyol.
The reaction is strongly dependent on the initial nucleophilic attack, which can be performed by a catalyst or impurities such as water [72][73]. Whereas in the industrial practice, epoxy/anhydride ratios ranging from 1:0.8 to 1:1 are common, only ratios as low as 1:0.5 are able to guarantee a reasonable supply of free –OH groups for transesterification [5][21][74]. As usual, the trade-off between maximised stiffness and vitrimeric properties arises. Among the most popular anhydrides used for vitrimeric networks, we mention: succinic anhydride [5][75][39], glutaric anhydride [19][73], Methylhexahydrophthalic Anhydride (MHHPA) [21], phthalic anhydride for a high-performance recyclable epoxy [76], as well as many others like maleic or trimellitic anhydride mentioned in several patents [77][37]. A very recent study by Giebler et al. showed that it is still possible to achieve a high Tg (140 °C) in an uncatalysed formulation of aminoglycidyl resin and glutaric anhydride [73]. The tertiary amine present in the epoxy monomer is able to catalyse the transesterification in the network, yet at a slower rate than Zn(Acac)2. The use of a technically-relevant resin and a non-toxic hardener gives this concept some concrete potential for industrial applications.
Epoxy/acid systems. Epoxy/acid is the prototypical vitrimeric system, dating back to the original study by Leibler et al. in 2011 [19]. The reaction mechanism is not straight-forward due to some possible side-reactions. However, the addition of carboxylic acid groups to epoxy rings yields β-hydroxy esters in stoichiometric conditions (Scheme 8), which are the key factor in transesterification [19][20][34]. Formulations based on fatty acids and epoxy monomers are suitable for varnishes and coatings, and the produced esters impart good adhesion, flexibility and water resistance [78]. Tran et al. envisioned a strategy to minimise hydrolysis in an epoxy/acid vitrimer produced by “miniemulsion” polymerisation [43]. The process involves the formation of an emulsion of pre-compatibilised precursors, followed by cross-linking, drying and sintering. Although this complex route requires careful optimisation (i.e., by the addition of a surfactant to stabilise the colloidal dispersion), the low viscosity and delayed sintering kinetics of the produced material might open new possibilities for the production of paints, coatings, as well as composite materials via RIM or RTM [43]. Han and Xu doped a DGEBA-citric acid network with different contents of polymethyl methacrylate, thus producing a polymer-polymer composite [79]. Contents up to 10–25 wt% gave rise to the formation of a network structure in the vitrimer, thereby reaching an optimum in tensile strength while also shortening the relaxation time [79]. As a final note, we mention Pripol® 1040, a mixture of di- and tri-carboxylic fatty acids used in combination with DGEBA in several studies focusing on vitrimers [80][14][36][42][81].
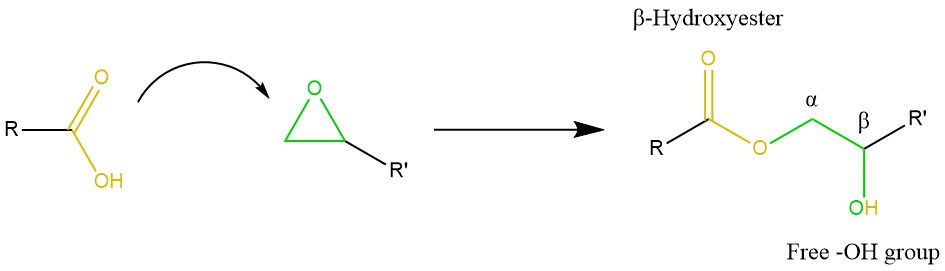
Scheme 8. Mechanism of epoxy/carboxylic acid curing. Usually with the aid of a catalyst, the acid binds the open oxirane ring, resulting in a β-hydroxy ester with a free –OH group. The latter is amenable to act as a nucleophile to initiate transesterification.
Epoxies with disulfide chemistry. The possibility of implementing a versatile chemistry such as disulfide chemistry in epoxies is definitely an intriguing option. In 1990, Tesoro and coworkers introduced an epoxy network cross-linked with a disulfide-containing diamine, showing the degradability and recyclability of the material by cleavage of S–S bonds [82]. By retracing these steps, another paper showed that a better degradability can be achieved by implementing disulfide bonds in the epoxy monomer instead [83]. Notably, disulfide metathesis can work in synergy with transesterification, as reported by Chen et al. in [84]. They introduced the concept of a dual vitrimer, where transesterification between DGEBA and dithiodibutyric acid is catalysed by TBD and disulfide exchange is triggered by heat. The material exhibited very fast stress relaxation (1.5 s at 200 °C with a very low estimated Tv) and excellent reprocessability after four cycles at 100 °C (1 h each). Lastly, as we have already brought to the reader’s attention, epoxies with disulfide chemistry may find applicability in FRPC [12].
3.2.3. Elastomers
Vulcanised rubbers quite arguably take a large share of the industrial market for elastomers, with more than 27 tons produced yearly, which represents a considerable hazard for the environment [85][33]. The most common attempts to reuse scrap elastomers consist of either grinding rubber waste into powder or chemical desulphurisation to reclaim the pre-vulcanized rubber network [33][41]. However, the original properties are irretrievably lost, thus leaving this option acceptable only in the case of less critical applications with low requirements [33]33. The interest in vitrimeric elastomers gravitates around these technological challenges. Here, we report studies on hydrogenated carboxylated nitrile-butadiene rubber [22], and composites thereof (study by our group under revision as of July 2020), polybutadiene/dioxaborolane networks for creep-resistant rubbers [85], carbon black-reinforced rubbers based on transesterification [41], recyclable sulphur-cured rubbers via transalkylation [33] and reprocessable epoxidised natural rubber via disulfide rearrangement [10].
Vitrimeric elastomers are also appealing for smart electronics, such as energy harvesters and tactile sensing elements [86]. Deng et al. proposed an elastomer amenable to disulfide exchange for encapsulating a conductive silver nanowire network [86]. The vitrimeric properties allowed for a facile and versatile fabrication of a triboelectric nanogenerator by multiple assembly of building blocks in a jigsaw puzzle-like fashion. The complex manipulation into freely-designable 3D structures with an origami strategy entails great potential for tactile sensors, with a possible extension to transistors in the foreseeable future [86].
Lastly, we report some studies on Polydimethylsiloxane (PDMS), a ubiquitous elastomer with potential in electronics (e.g., as an ion-selective membrane in Ion-Sensitive Field-Effect Transistor (ISFETs), spring material in accelerometers or in sensors with integrated electronics) [87]. Zheng and McCarthy revived the attention for siloxane equilibration as a “simple, robust and obvious” self-healing mechanism, possibly overlooked ever since the first publications from the 1950s [74]. They designed a living polymer network from the equilibrium of cyclosiloxane oligomers and tetramethylammonium dimethylsilanolate. Stability to hydrolysis, catalyst-free formulation and a low-cost monomer are the major advantages. Schmolke et al. tried to apply this concept to a room-temperature self-healing PDMS [11]. Lastly, Stukenbroeker and coworkers reported a quenchable PDMS vitrimeric network with dynamic vinylogous urethane cross-links [88]. Stress relaxation could be both activated thermally and suppressed on demand by reacting a -ketoester with the free -NH2 groups. This “masking” effect eventually impedes transamination, hence leading to a quenched state [88].
3.2.4. Composites and Nanocomposites
As customary in industrial practice, polymeric matrices are usually reinforced by means of inorganic fillers to boost characteristics such as thermomechanical properties, wear performance and environmental resistance. When the filler comes in the form of small solid particles with dimensions below 100 nm, these materials are referred to as nanocomposites. Krishnakumar et al. used Graphene Oxide (GO) nanofillers in an epoxy vitrimer that underwent network rearrangement by disulfide exchange [89]. Intriguingly, a higher GO content generates additional free volume, thus lowering Tg and enhancing chain mobility. As a consequence of this, self-healing and shape memory properties are more favourable than in unfilled matrices. This effect, together with the improved flexural strength and modulus (+7.1% and +9.4% for 1 wt% GO, respectively), contributes to the attractiveness of the formulation [89].
However, this approach takes advantage of the poor interaction between GO and the organic matrix. On the contrary, a good adhesion between filler and matrix is desirable in composites, in order to assure a correct load transfer. Legrand and coworkers designed nanocomposites based on epoxy-functionalised silica nanoparticles [14]. The grafted chains limit filler aggregation by steric hindrance and favour a good dispersion state. Although unfunctionalised particles showed the tendency to constrain chain movement, the active β-hydroxy esters directly bonded onto the surface improve stress relaxation substantially [14]. The authors then proceeded with the production of a large batch of nanocomposite formulation (25 wt% of industrially-relevant silica particles), proving the flexibility of their design. In an unpublished study by our group (under revision as of July 2020), we proposed a similar approach for composites from Hydrogenated carboxylated Nitrile-Butadiene Rubber (HXNBR) and epoxy-functionalised calcium silicate particles (esilicate). The urge for rubber reinforcement with a high filler content rises from the poor mechanical performance of the raw elastomer, and it is therefore tremendously desired for industrial applications. In our unpublished study, we used the functionalised particles both as the only source of exchangeable bonds and as the cross-linker. Because of this, the TBD-catalysed transesterification became faster as the amount of the filler increased. This unusual advantage came together with a mild plasticising effect, which led to a decrease in Tg with the inclusion of more esilicate particles. The incomplete stress-relaxation makes this material more appropriately regarded as a vitrimer-like composite.
Another clever concept was proposed by Yang et al. in [45]. They produced a graphene-reinforced resin based on TBD, DGEBA and sebacic acid. The shape memory effect allowed switching from a 2D shape to a 3D one via a twofold approach: besides the standard thermally-triggered transesterification, graphene enabled energy conversion upon Near-Infrared Radiation (NIR). That is to say, graphene interacts with light to produce heat (which goes by the name of the photothermal effect), thus activating network rearrangement indirectly. This effect allowed shape recovery in roughly 1 min. Moreover, as the graphene loading stepped from 0% to 1 wt%, the material showed ductile fracture, with larger yield strength and strain at break [45]. The same dual-triggered approach was used by Yan et al. in a vitrimeric polyurethane reinforced with carbon-nanotubes [90]. The response to NIR enabled shape recovery and reshaping under heat and light with no mould, thanks to the accelerated transcarbamoylation.
On a different note, Yu et al. tried to improve the production protocol of compression moulding for long fibre composites, with the introduction of a vitrimeric powder [81]. The vitrimer particles become cured and sintered, while at the same time reinforced with a carbon fibre mat in one step. Since there is no precuring stage for the matrix, the procedure could be completed in only 1 min (1 MPa at 300 °C) [81]. The malleability was demonstrated by reshaping the produced sheet into a pinwheel and by subsequently heating up at 200 °C for 3 h. Other relevant studies on composites have been covered in the other sections above [91][12][92][46][53].
4. Outlook and Conclusions
In summary, our work offers a comprehensive overview of CANs as a potent and fresh resource in polymer science and engineering. With the increasing concern for environmental issues and hazardous chemical waste, the possibility of extending the service of thermosetting polymers via self-healing and recycling is undoubtedly intriguing. As presented above, this option is viable for applications in the aerospace, automotive, electronics and biomedical sectors, as far as composites, sustainable thermosets from bio-renewable sources, epoxies, elastomers and polyurethanes are concerned. Vitrimers, introduced to the scientific community no longer than nine years ago, are a budding class of thermosets with the ability to flow above a certain characteristic temperature determined by the chemistry involved. This property is achieved by covalent bond exchange, for instance via transesterification, transamination or disulfide metathesis. The underlying concept of vitrimers is based on the relatively simple idea of a network amenable to self-arrange upon stimulation, ideally with no loss of integrity and no undesired side-reactions over time. Therein lies their potential. However, the current dearth of practical applicability in industry may be ascribable to some inherent limitations. Firstly, to the best of our knowledge, the trade-off between self-healing (or recycling) efficiency and mechanical performance still represents a major scientific conundrum. Secondly, the typical long relaxation times observed in vitrimeric systems are far away from meeting the standards of conventional processing routes for thermoplastics, where the molten polymer is shaped in a few seconds. On the contrary, the applicability in composite processing seems much more promising, albeit still vastly underexplored. Further drawbacks are the lack of a well-established protocol for gauging Tv, as well as the shortage of fit-for-purpose, sustainable formulations. In view of the above-mentioned challenges, we are thrilled to be contributing further to the improvement of vitrimers. We are confident that a bright future is ahead for the applicability of CANs in industry.
References
- Pleşa, I.; Noţingher, P.V.; Schlögl, S.; Sumereder, C.; Muhr, M. Properties of Polymer Composites Used in High-Voltage Applications. Polymers 2016, 8, 173.
- Pleşa, I.; Noţingher, P.V.; Stancu, C.; Wiesbrock, F.; Schlögl, S. Polyethylene Nanocomposites for Power Cable Insulations. Polymers 2018, 11, 24.
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108.
- Zou, W.; Dong, J.; Luo, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29, 1606100.
- Liu, T.; Hao, C.; Wang, L.; Li, Y.; Liu, W.; Xin, J.; Zhang, J. Eugenol-Derived Biobased Epoxy: Shape Memory, Repairing, and Recyclability. Macromolecules 2017, 50, 8588–8597.
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38.
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173.
- Lu, L.; Fan, J.; Li, G. Intrinsic healable and recyclable thermoset epoxy based on shape memory effect and transesterification reaction. Polymer 2016, 105, 10–18.
- Byrne, J.P. Rubber Elasticity: A Simple Method for Measurement of Thermodynamic Properties. J. Chem. Educ. 1994, 71, 531.
- Imbernon, L.; Oikonomou, E.K.; Norvez, S.; Leibler, L. Chemically cross-linked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements. Polym. Chem. 2015, 6, 4271–4278.
- Schmolke, W.; Perner, N.; Seiffert, S. Dynamically Cross-Linked Polydimethylsiloxane Networks with Ambient-Temperature Self-Healing. Macromolecules 2015, 48, 8781–8788.
- Ruiz de Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide cross-links to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horizons 2016, 3, 241–247.
- Röttger, M.; Domenech, T.; van der Weegen, R.; Breuillac, A.; Nicolaÿ, R.; Leibler, L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356, 62–65.
- Legrand, A.; Soulié-Ziakovic, C. Silica–Epoxy Vitrimer Nanocomposites. Macromolecules 2016, 49, 5893–5902.
- Obadia, M.M.; Mudraboyina, B.P.; Serghei, A.; Montarnal, D.; Drockenmuller, E. Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C-N Bonds. J. Am. Chem. Soc. 2015, 137, 6078–6083.
- Self, J.L.; Dolinski, N.D.; Zayas, M.S.; Read de Alaniz, J.; Bates, C.M. Brønsted-Acid-Catalyzed Exchange in Polyester Dynamic Covalent Networks. ACS Macro Lett. 2018, 7, 817–821.
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470.
- Liu, T.; Zhao, B.; Zhang, J. Recent development of repairable, malleable and recyclable thermosetting polymers through dynamic transesterification. Polymer 2020, 194, 122392.
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968.
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667.
- Liu, T.; Hao, C.; Zhang, S.; Yang, X.; Wang, L.; Han, J.; Li, Y.; Xin, J.; Zhang, J. A Self-Healable High Glass Transition Temperature Bioepoxy Material Based on Vitrimer Chemistry. Macromolecules 2018, 51, 5577–5585.
- Kaiser, S.; Wurzer, S.; Pilz, G.; Kern, W.; Schlögl, S. Stress relaxation and thermally adaptable properties in vitrimer-like elastomers from HXNBR rubber with covalent bonds. Soft Matter 2019, 15, 6062–6072.
- Taplan, C.; Guerre, M.; Winne, J.M.; Du Prez, F.E. Fast processing of highly cross-linked, low-viscosity vitrimers. Mater. Horizons 2020, 7, 104–110.
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457.
- Denissen, W.; de Baere, I.; van Paepegem, W.; Leibler, L.; Winne, J.; Du Prez, F.E. Vinylogous Urea Vitrimers and Their Application in Fiber Reinforced Composites. Macromolecules 2018, 51, 2054–2064.
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically activated, catalyst-free polyhydroxyurethane vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022.
- Zheng, N.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Thermoset Shape-Memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem. 2016, 55, 11421–11425.
- Zheng, N.; Hou, J.; Xu, Y.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Catalyst-Free Thermoset Polyurethane with Permanent Shape Reconfigurability and Highly Tunable Triple-Shape Memory Performance. ACS Macro Lett. 2017, 6, 326–330.
- Chen, X.; Li, L.; Jin, K.; Torkelson, J.M. Reprocessable polyhydroxyurethane networks exhibiting full property recovery and concurrent associative and dissociative dynamic chemistry via transcarbamoylation and reversible cyclic carbonate aminolysis. Polym. Chem. 2017, 8, 6349–6355.
- Jourdain, A.; Asbai, R.; Anaya, O.; Chehimi, M.M.; Drockenmuller, E.; Montarnal, D. Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks. Macromolecules 2020, 53, 1884–1900.
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Structural effects on the reprocessability and stress relaxation of cross-linked polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 44984.
- Chakma, P.; Konkolewicz, D. Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem. Int. Ed. 2019, 58, 9682–9695.
- Tang, Z.; Liu, Y.; Huang, Q.; Zhao, J.; Guo, B.; Zhang, L. A real recycling loop of sulphur-cured rubber through transalkylation exchange of C–S bonds. Green Chem. 2018, 20, 5454–5458.
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1, 789–792.
- Lossada, F.; Guo, J.; Jiao, D.; Groeer, S.; Bourgeat-Lami, E.; Montarnal, D.; Walther, A. Vitrimer Chemistry Meets Cellulose Nanofibrils: Bioinspired Nanopapers with High Water Resistance and Strong Adhesion. Biomacromolecules 2019, 20, 1045–1055.
- Altuna, F.I.; Hoppe, C.E.; Williams, R.J.J. Shape memory epoxy vitrimers based on DGEBA cross-linked with dicarboxylic acids and their blends with citric acid. RSC Adv. 2016, 6, 88647–88655.
- Duquenne, C.; Melas, M.; Gentilhomme, P.; Disson, J.-P. Composition for Manufacturing Epoxy/Anhydride Vitrimer Resins including an Organic Catalyst. US 20170044305A1, 16 February 2017.
- DelDonno, T.A. Polyurethane Preparation Using Organo-Zinc Catalyst and Time-Lapse Modifier. U.S. Patent 4,426,510, 17 January 1984.
- Han, J.; Liu, T.; Zhang, S.; Hao, C.; Xin, J.; Guo, B.; Zhang, J. Hyperbranched Polymer Assisted Curing and Repairing of an Epoxy Coating. Ind. Eng. Chem. Res. 2019, 58, 6466–6475.
- Zhang, B.; Kowsari, K.; Serjouei, A.; Dunn, M.L.; Ge, Q. Reprocessable thermosets for sustainable three-dimensional printing. Nat. Commun. 2018, 9, 1831.
- Zhang, G.; Zhou, X.; Liang, K.; Guo, B.; Li, X.; Wang, Z.; Zhang, L. Mechanically Robust and Recyclable EPDM Rubber Composites by a Green Cross-Linking Strategy. ACS Sustain. Chem. Eng. 2019, 7, 11712–11720.
- Demongeot, A.; Mougnier, S.J.; Okada, S.; Soulié-Ziakovic, C.; Tournilhac, F. Coordination and catalysis of Zn2+ in epoxy-based vitrimers. Polym. Chem. 2016, 7, 4486–4493.
- Tran, T.N.; Rawstron, E.; Bourgeat-Lami, E.; Montarnal, D. Formation of Cross-Linked Films from Immiscible Precursors through Sintering of Vitrimer Nanoparticles. ACS Macro Lett. 2018, 7, 376–380.
- Niu, X.; Wang, F.; Li, X.; Zhang, R.; Wu, Q.; Sun, P. Using Zn2+ Ionomer To Catalyze Transesterification Reaction in Epoxy Vitrimer. Ind. Eng. Chem. Res. 2019, 58, 5698–5706.
- Yang, Z.; Wang, Q.; Wang, T. Dual-Triggered and Thermally Reconfigurable Shape Memory Graphene-Vitrimer Composites. ACS Appl. Mater. Interfaces 2016, 8, 21691–21699.
- Yang, Y.; Pei, Z.; Zhang, X.; Tao, L.; Wei, Y.; Ji, Y. Carbon nanotube–vitrimer composite for facile and efficient photo-welding of epoxy. Chem. Sci. 2014, 5, 3486–3492.
- Kiesewetter, M.K.; Scholten, M.D.; Kirn, N.; Weber, R.L.; Hedrick, J.L.; Waymouth, R.M. Cyclic guanidine organic Catalysts: What is magic about triazabicyclodecene? J. Org. Chem. 2009, 74, 9490–9496.
- Yadav, J.S.; Reddy, B.; Krishna, A.D.; Reddy, C.S.; Narsaiah, A.V. Triphenylphosphine: An efficient catalyst for transesterification of β-ketoesters. J. Mol. Catal. A Chem. 2007, 261, 93–97.
- Brutman, J.P.; Delgado, P.A.; Hillmyer, M.A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610.
- Lou, C.; Gu, J.; Di, M.; Ma, L.; Wang, Y.; Liu, X. Synthesis and Characterization of Trichlorophenol-blocked Polyaryl Polyisocyanate. Iran. Polym. J. 2011, 20, 247–255.
- Liu, W.; Schmidt, D.F.; Reynaud, E. Catalyst Selection, Creep, and Stress Relaxation in High-Performance Epoxy Vitrimers. Ind. Eng. Chem. Res. 2017, 56, 2667–2672.
- Snyder, R.L.; Fortman, D.J.; de Hoe, G.X.; Hillmyer, M.A.; Dichtel, W.R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51, 389–397.
- Zhao, W.; Feng, Z.; Liang, Z.; Lv, Y.; Xiang, F.; Xiong, C.; Duan, C.; Dai, L.; Ni, Y. Vitrimer-Cellulose Paper Composites: A New Class of Strong, Smart, Green, and Sustainable Materials. ACS Appl. Mater. Interfaces 2019, 11, 36090–36099.
- Hatano, M.; Tabata, Y.; Yoshida, Y.; Toh, K.; Yamashita, K.; Ogura, Y.; Ishihara, K. Metal-free transesterification catalyzed by tetramethylammonium methyl carbonate. Green Chem. 2018, 20, 1193–1198.
- Rivero, G.; Nguyen, L.T.T.; Hillewaere, X.K.D.; Du Prez, F.E. One-Pot Thermo-Remendable Shape Memory Polyurethanes. Macromolecules 2014, 47, 2010–2018.
- Billiet, S.; de Bruycker, K.; Driessen, F.; Goossens, H.; van Speybroeck, V.; Winne, J.M.; Du Prez, F.E. Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems. Nat. Chem. 2014, 6, 815–821.
- Roling, O.; de Bruycker, K.; Vonhören, B.; Stricker, L.; Körsgen, M.; Arlinghaus, H.F.; Ravoo, B.J.; Du Prez, F.E. Rewritable Polymer Brush Micropatterns Grafted by Triazolinedione Click Chemistry. Angew. Chem. Int. Ed. 2015, 54, 13126–13129.
- Zhang, S.; Liu, T.; Hao, C.; Wang, L.; Han, J.; Liu, H.; Zhang, J. Preparation of a lignin-based vitrimer material and its potential use for recoverable adhesives. Green Chem. 2018, 20, 2995–3000.
- Zhang, W.; Ma, Y.; Wang, C.; Li, S.; Zhang, M.; Chu, F. Preparation and properties of lignin–phenol– formaldehyde resins based on different biorefinery residues of agricultural biomass. Ind. Crops Prod. 2013, 43, 326–333.
- Hernandez, E.D.; Bassett, A.W.; Sadler, J.M.; La Scala, J.J.; Stanzione, J.F. Synthesis and Characterization of Bio-based Epoxy Resins Derived from Vanillyl Alcohol. ACS Sustain. Chem. Eng. 2016, 4, 4328–4339.
- Effendi, A.; Gerhauser, H.; Bridgwater, A.V. Production of renewable phenolic resins by thermochemical conversion of biomass: A review. Renew. Sustain. Energy Rev. 2008, 12, 2092–2116.
- Foyer, G.; Chanfi, B.H.; Boutevin, B.; Caillol, S.; David, G. New method for the synthesis of formaldehyde-free phenolic resins from lignin-based aldehyde precursors. Eur. Polym. J. 2016, 74, 296–309.
- Xin, J.; Li, M.; Li, R.; Wolcott, M.P.; Zhang, J. Green Epoxy Resin System Based on Lignin and Tung Oil and Its Application in Epoxy Asphalt. ACS Sustain. Chem. Eng. 2016, 4, 2754–2761.
- Cantarutti, C.; Dinu, R.; Mija, A. Biorefinery Byproducts and Epoxy Biorenewable Monomers: A Structural Elucidation of Humins and Triglycidyl Ether of Phloroglucinol Cross-Linking. Biomacromolecules 2020, 21, 517–533.
- Geng, H.; Wang, Y.; Yu, Q.; Gu, S.; Zhou, Y.; Xu, W.; Zhang, X.; Ye, D. Vanillin-Based Polyschiff Vitrimers: Reprocessability and Chemical Recyclability. ACS Sustain. Chem. Eng. 2018, 6, 15463–15470.
- Dhers, S.; Vantomme, G.; Avérous, L. A fully bio-based polyimine vitrimer derived from fructose. Green Chem. 2019, 21, 1596–1601.
- Yan, N.; Chen, X. Sustainability: Don’t waste seafood waste. Nature 2015, 524, 155–157.
- Zhang, J.; Feng, M.; Lu, X.; Shi, C.; Li, X.; Xin, J.; Yue, G.; Zhang, S. Base-free preparation of low molecular weight chitin from crab shell. Carbohydr. Polym. 2018, 190, 148–155.
- Anitha, A.; Sowmya, S.; Kumar, P.S.; Deepthi, S.; Chennazhi, K.P.; Ehrlich, H.; Tsurkan, M.; Jayakumar, R. Chitin and chitosan in selected biomedical applications. Prog. Polym. Sci. 2014, 39, 1644–1667.
- Ghosh, B.; Urban, M.W. Self-repairing oxetane-substituted chitosan polyurethane networks. Science 2009, 323, 1458–1460.
- Boey, F.; Qiang, W. Experimental modeling of the cure kinetics of an epoxy-hexaanhydro-4-methylphthalicanhydride (MHHPA) system. Polymer 2000, 41, 2081–2094.
- Paramarta, A.; Webster, D.C. Curing kinetics of bio-based epoxy-anhydride thermosets with zinc catalyst. J. Therm. Anal. Calorim. 2017, 130, 2133–2144.
- Giebler, M.; Sperling, C.; Kaiser, S.; Duretek, I.; Schlögl, S. Epoxy-Anhydride Vitrimers from Aminoglycidyl Resins with High Glass Transition Temperature and Efficient Stress Relaxation. Polymers 2020, 12, 1148.
- Zheng, P.; McCarthy, T.J. A surprise from 1954: Siloxane equilibration is a simple, robust, and obvious polymer self-healing mechanism. J. Am. Chem. Soc. 2012, 134, 2024–2027.
- Han, J.; Liu, T.; Hao, C.; Zhang, S.; Guo, B.; Zhang, J. A Catalyst-Free Epoxy Vitrimer System Based on Multifunctional Hyperbranched Polymer. Macromolecules 2018, 51, 6789–6799.
- Lu, L.; Pan, J.; Li, G. Recyclable high-performance epoxy based on transesterification reaction. J. Mater. Chem. A 2017, 5, 21505–21513.
- Duquenne, C.; Melas, M.; Beaudrais, S. Composition for Manufacturing Vitrimer Resins of Epoxy/anhydride Type Comprising a Polyol. U.S. Patent 2017/0044361 A1, 16 February 2017.
- Altuna, F.I.; Hoppe, C.E.; Williams, R.J.J. Epoxy Vitrimers: The Effect of Transesterification Reactions on the Network Structure. Polymers 2018, 10, 43.
- Han, H.; Xu, X. Poly(methyl methacrylate)-epoxy vitrimer composites. J. Appl. Polym. Sci. 2018, 135, 46307.
- Shi, Q.; Yu, K.; Kuang, X.; Mu, X.; Dunn, C.K.; Dunn, M.L.; Wang, T.; Jerry Qi, H. Recyclable 3D printing of vitrimer epoxy. Mater. Horizons 2017, 4, 598–607.
- Yu, L.; Zhu, C.; Sun, X.; Salter, J.; Wu, H.; Jin, Y.; Zhang, W.; Long, R. Rapid Fabrication of Malleable Fiber Reinforced Composites with Vitrimer Powder. ACS Appl. Polym. Mater. 2019, 1, 2535–2542.
- Tesoro, G.C.; Sastri, V. Reversible cross-linking in epoxy resins. I. Feasibility studies. J. Appl. Polym. Sci. 1990, 39, 1425–1437.
- Takahashi, A.; Ohishi, T.; Goseki, R.; Otsuka, H. Degradable epoxy resins prepared from diepoxide monomer with dynamic covalent disulfide linkage. Polymer 2016, 82, 319–326.
- Chen, M.; Zhou, L.; Wu, Y.; Zhao, X.; Zhang, Y. Rapid Stress Relaxation and Moderate Temperature of Malleability Enabled by the Synergy of Disulfide Metathesis and Carboxylate Transesterification in Epoxy Vitrimers. ACS Macro Lett. 2019, 8, 255–260.
- Breuillac, A.; Kassalias, A.; Nicolaÿ, R. Polybutadiene Vitrimers Based on Dioxaborolane Chemistry and Dual Networks with Static and Dynamic Cross-links. Macromolecules 2019, 52, 7102–7113.
- Deng, J.; Kuang, X.; Liu, R.; Ding, W.; Wang, A.C.; Lai, Y.C.; Dong, K.; Wen, Z.; Wang, Y.; Wang, L.; et al. Vitrimer Elastomer-Based Jigsaw Puzzle-Like Healable Triboelectric Nanogenerator for Self-Powered Wearable Electronics. Adv. Mater. 2018, 30, e1705918.
- Lötters, J.C.; Olthuis, W.; Veltink, P.H.; Bergveld, P. The mechanical properties of the rubber elastic polymer polydimethylsiloxane for sensor applications. J. Micromech. Microeng. 1997, 7, 145–147.
- Stukenbroeker, T.; Wang, W.; Winne, J.M.; Du Prez, F.E.; Nicolaÿ, R.; Leibler, L. Polydimethylsiloxane quenchable vitrimers. Polym. Chem. 2017, 8, 6590–6593.
- Krishnakumar, B.; Prasanna Sanka, R.; Binder, W.H.; Park, C.; Jung, J.; Parthasarthy, V.; Rana, S.; Yun, G.J. Catalyst free self-healable vitrimer/graphene oxide nanocomposites. Compos. Part B Eng. 2020, 184, 107647.
- Yan, P.; Zhao, W.; Jiang, L.; Wu, B.; Hu, K.; Yuan, Y.; Lei, J. Reconfiguration and shape memory triggered by heat and light of carbon nanotube-polyurethane vitrimer composites. J. Appl. Polym. Sci. 2018, 135, 45784.
- Kuang, X.; Zhou, Y.; Shi, Q.; Wang, T.; Qi, H.J. Recycling of Epoxy Thermoset and Composites via Good Solvent Assisted and Small Molecules Participated Exchange Reactions. ACS Sustain. Chem. Eng. 2018, 6, 9189–9197.
- Chabert, E.; Vial, J.; Cauchois, J.P.; Mihaluta, M.; Tournilhac, F. Multiple welding of long fiber epoxy vitrimer composites. Soft Matter 2016, 12, 4838–4845.



