 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Guilherme Ferreira | + 4540 word(s) | 4540 | 2020-07-23 10:54:08 | | | |
| 2 | Rita Xu | -720 word(s) | 3820 | 2020-07-31 03:59:23 | | |
Video Upload Options
Reports of a reproducibility crisis combined with a high attrition rate in the pharmaceutical industry have put animal research increasingly under scrutiny in the past decade. Many researchers and the general public now question whether there is still a justification for conducting animal studies. While criticism of the current modus operandi in preclinical research is certainly warranted, the data on which these discussions are based are often unreliable. Several initiatives to address the internal validity and reporting quality of animal studies (e.g., Animals in Research: Reporting In Vivo Experiments (ARRIVE) and Planning Research and Experimental Procedures on Animals: Recommendations for Excellence (PREPARE) guidelines) have been introduced but seldom implemented. As for external validity, progress has been virtually absent. Nonetheless, the selection of optimal animal models of disease may prevent the conducting of clinical trials, based on unreliable preclinical data. Here, we discuss three contributions to tackle the evaluation of the predictive value of animal models of disease themselves. First, we developed the Framework to Identify Models of Disease (FIMD), the first step to standardise the assessment, validation and comparison of disease models. FIMD allows the identification of which aspects of the human disease are replicated in the animals, facilitating the selection of disease models more likely to predict human response. Second, we show an example of how systematic reviews and meta-analyses can provide another strategy to discriminate between disease models quantitatively. Third, we explore whether external validity is a factor in animal model selection in the Investigator’s Brochure (IB), and we use the IB-derisk tool to integrate preclinical pharmacokinetic and pharmacodynamic data in early clinical development. Through these contributions, we show how we can address external validity to evaluate the translatability and scientific value of animal models in drug development. However, while these methods have potential, it is the extent of their adoption by the scientific community that will define their impact. By promoting and adopting high-quality study design and reporting, as well as a thorough assessment of the translatability of drug efficacy of animal models of disease, we will have robust data to challenge and improve the current animal research paradigm.
1. Definition
Reports of a reproducibility crisis combined with a high attrition rate in the pharmaceutical industry have put animal research increasingly under scrutiny in the past decade. Many researchers and the general public now question whether there is still a justification for conducting animal studies. While criticism of the current modus operandi in preclinical research is certainly warranted, the data on which these discussions are based are often unreliable. Several initiatives to address the internal validity and reporting quality of animal studies (e.g., Animals in Research: Reporting In Vivo Experiments (ARRIVE) and Planning Research and Experimental Procedures on Animals: Recommendations for Excellence (PREPARE) guidelines) have been introduced but seldom implemented. As for external validity, progress has been virtually absent. Nonetheless, the selection of optimal animal models of disease may prevent the conducting of clinical trials, based on unreliable preclinical data.
2. Introduction
Despite modest improvements, the attrition rate in the pharmaceutical industry remains high [1][2][3]. Although the explanation for such low success is multifactorial, the lack of translatability of animal research has been touted as a critical aspect [4][5]. Evidence of the problems of animal research’s modus operandi has been mounting for almost a decade. Research has shown preclinical studies have major design flaws (e.g., low power, irrelevant endpoints), are poorly reported, or both—leading to unreliable data which ultimately means that animals are subjected to unnecessary suffering and clinical trial participants are potentially placed at risk [6][7]. Increasing reports of failure to reproduce preclinical studies across several fields pointed to the need to increase current standards [8][9][10]. In these terms, the assessment of two essential properties of translational research—internal and external validity—is jeopardised [11].
Internal validity refers to whether the findings of an experiment in defined conditions are true [4]. Measures related to internal validity, such as randomisation and blinding, reduce or prevent several types of bias, and have a profound effect on study outcomes [12][13]. Initiatives, such as the Animals in Research: Reporting In Vivo Experiments (ARRIVE) and Planning Research and Experimental Procedures on Animals: Recommendations for Excellence (PREPARE) guidelines and harmonized animal research reporting principles (HARRP), address the issues related to the poor design and reporting of animal experiments [14][15][16]. While all of these initiatives can resolve most, if not all, issues surrounding internal validity, they are poorly implemented and their uptake by all stakeholders is remarkably slow [13][17].
If progress on the internal validity front has been insufficient, for external validity, it has been virtually absent. External validity is related to whether an experiment’s findings can be extrapolated to other circumstances (e.g., animal to human translation). For external validity, while the study design plays a significant role (e.g., relevant endpoints and time to treatment), there is another often overlooked dimension—the animal models themselves [18]. The pitfalls of using animals to simulate human conditions, such as different aetiology and lack of genetic heterogeneity, have been widely recognised for a long time [1][10][19][20]. Nonetheless, the few efforts to address external validity to the same extent as internal validity are still insufficient [21][22].
The results of a sizeable portion of animal studies are unreliable [8][9][23]. If we cannot fully trust the data generated by animal experiments, how can we assess their value? We argue that for the sensible evaluation of animal models of disease, we need to generate robust data first. To generate robust data, we need models that simulate the human disease features to the extent that allows for reliable translation between species. Through the selection of optimal animal models of disease, we can potentially prevent clinical trials from advancing based on data unlikely to translate.
2. The Framework to Identify Models of Disease (FIMD)
The evaluation of preclinical efficacy often employs animal models of disease. Here, we use ‘animal model of disease’ for any animal model that simulates a human condition (or symptom) for which a drug can be developed, including testing paradigms. While safety studies are tightly regulated, including standardisation of species and study designs, there is hardly any guidance available for efficacy [24]. New drugs often have new and unknown mechanisms of action, which require tailor-made approaches for their elucidation [24][25]. As such, it would be counterproductive for regulatory agencies and companies alike to predetermine models or designs for efficacy as it is done for safety. However, in practice, this lack of guidance has contributed to the performance of studies with considerable methodological flaws [6][26].
The assessment of the external validity of animal models has traditionally relied on the criteria of face, construct and predictive validity [27][28]. These concepts are generic and highly prone to user interpretation, leading to the analysis of disease models according to different disease parameters. This situation complicates an objective comparison between animal models. Newer approaches, such as the tool developed by Sams-Dodd and Denayer et al., can be applied to in vitro and in vivo models and consist of the simple scoring of five categories (species, disease simulation, face validity, complexity and predictivity) according to their proximity to the human condition [20][29]. Nevertheless, they still fail to capture relevant characteristics involved in the pathophysiology and drug response, such as histology and biomarkers.
To address the lack of standardisation and the necessity of a multidimensional appraisal of animal models of disease, we developed the Framework to Identify Models of Disease (FIMD) [21][22]. The first step in the development of FIMD was the identification of the core parameters used to validate disease models in the literature. Eight domains were identified: Epidemiology, Symptomatology and Natural History (SNH), Genetics, Biochemistry, Aetiology, Histology, Pharmacology and Endpoints. More than 60% of the papers included in our scoping review used three or fewer domains. As it stood, the validation of animal models followed the tendency of the field as a whole—no standardisation nor integration of the characteristics frequently mentioned as relevant.
Based on these results, we drafted questions about each domain to determine the similarity to the human condition (Table 1). The sheet containing the answers to these questions and references thereof is called a validation sheet. The weighting and scoring system weighs all domains equally. The final score can be visualised in a radar plot of the eight domains and, together with the validation sheet, facilitates the comparison of disease models at a high level. An example of a radar plot is presented in Figure 1.
Figure 1. Example of a radar plot obtained with the validation of two animal models using the Framework to Identify Models of Disease (FIMD). SNH—Symptomatology and Natural History. Extracted from Ferreira et al. [21][22].
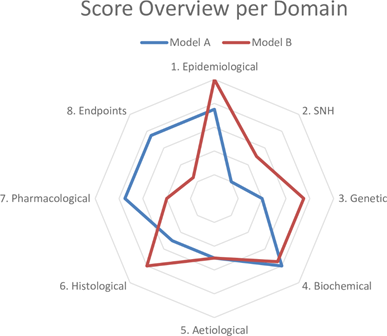
Table 1. Questions per domain with weighting per questions. Extracted from Ferreira et al. [21][22].
|
|
Weight |
|
1. EPIDEMIOLOGICAL VALIDATION |
|
|
1.1 Is the model able to simulate the disease in the relevant sexes? |
6.25 |
|
1.2 Is the model able to simulate the disease in the relevant age groups (e.g., juvenile, adult or ageing)? |
6.25 |
|
2. SYMPTOMATOLOGY AND NATURAL HISTORY VALIDATION |
12.5 |
|
2.1 Is the model able to replicate the symptoms and co-morbidities commonly present in this disease? If so, which ones? |
2.5 |
|
2.2 Is the natural history of the disease similar to human’s regarding: 2.2.1 Time to onset |
2.5 |
|
2.2.2 Disease progression |
2.5 |
|
2.2.3 Duration of symptoms |
2.5 |
|
2.2.4 Severity |
2.5 |
|
3. GENETIC VALIDATION |
12.5 |
|
3.1 Does this species also have orthologous genes and/or proteins involved in the human disease? |
4.17 |
|
3.2 If so, are the relevant genetic mutations or alterations also present in the orthologous genes/proteins? |
4.17 |
|
3.3 If so, is the expression of such orthologous genes and/or proteins similar to the human condition? |
4.16 |
|
4. BIOCHEMICAL VALIDATION |
12.5 |
|
4.1 If there are known pharmacodynamic (PD) biomarkers related to the pathophysiology of the disease, are they also present in the model? |
3.125 |
|
4.2 Do these PD biomarkers behave similarly to humans’? |
3.125 |
|
4.3 If there are known prognostic biomarkers related to the pathophysiology of the disease, are they also present in the model? |
3.125 |
|
4.4 Do these prognostic biomarkers behave similarly to humans’? |
3.125 |
|
5. AETIOLOGICAL VALIDATION |
12.5 |
|
5.1 Is the aetiology of the disease similar to humans’? |
12.5 |
|
6. HISTOLOGICAL VALIDATION |
12.5 |
|
6.1 Do the histopathological structures in relevant tissues resemble the ones found in humans? |
12.5 |
|
7. PHARMACOLOGICAL VALIDATION |
12.5 |
|
7.1 Are effective drugs in humans also effective in this model? |
4.17 |
|
7.2 Are ineffective drugs in humans also ineffective in this model? |
4.17 |
|
7.3 Have drugs with different mechanisms of action and acting on different pathways been tested in this model? If so, which? |
4.16 |
|
8. ENDPOINT VALIDATION |
12.5 |
|
8.1 Are the endpoints used in preclinical studies the same or translatable to the clinical endpoints? |
6.25 |
|
8.2 Are the methods used to assess preclinical endpoints comparable to the ones used to assess related clinical endpoints? |
6.25 |
To account for the low internal validity of animal research, we added a reporting quality and risk of bias assessment for the pharmacological validation section. This section includes all studies in which a drug intervention was tested. The reporting quality parameters were based on the ARRIVE guidelines, and the risk of bias questions were extracted from the tool published by the SYstematic Review Center for Laboratory animal Experimentation (SYRCLE) [14][30]. With this information, researchers can put pharmacological studies into context and evaluate how reliable the results are likely to be.
The final contribution of FIMD was a validation definition for animal models of disease. We grounded the definition of validation on the evidence provided for a model’s context of use, grading it into four levels of confidence. With this definition, we intentionally decoupled the connotation of a validated model being a predictive model. Rather, we reinforce that a validated animal model is a model with well-defined, reproducible characteristics.
To validate our framework, we first conducted a pilot study of two models of type 2 diabetes—the Zucker Diabetic Fatty (ZDF) rat and db/db mouse—chosen on the basis of their extensive use in preclinical studies. Next, we did a complete validation of two models of Duchenne Muscular Dystrophy (DMD)—the mdx mouse and the Golden Retriever Muscular Dystrophy (GRMD) dog. We chose the mdx mouse owing to its common use as a DMD model and the GRMD dog for its similarities to the human condition [31][32]. While only minor differences were found for the type 2 diabetes models, the models for DMD presented more striking dissimilarities. The GRMD dog scored higher in the Epidemiology, SNH and Histology domains, whereas the mdx mouse did so in the Pharmacology and Endpoints domains, the latter mainly driven by the absence of studies in dogs. Our findings indicate that the mdx mouse may not be appropriate to test disease-modifying drugs, despite its use in most animal studies in DMD [31]. If more pharmacology studies are published using the GRMD dog, it will result in a more refined assessment. A common finding in all the four models was the high prevalence of experiments for which the risk of bias could not be assessed.
We designed FIMD to avoid the limitations of previous approaches, which included the lack of and external validities. Nonetheless, it presents challenges of its own, ranging from the definition of disease parameters and absence of a statistical model to support a more sensitive weighting and scoring system to the use of publicly available (and often biased) data. The latter is especially relevant, as study design and reporting deficiencies of animal studies undoubtedly represent a challenge to interpret the resulting data correctly. While owing to these deficiencies, many studies are less informative; some may still offer potential insights if the data are interpreted adequately. We included the reporting quality and risk of bias assessment to force researchers to account for these shortcomings when interpreting the data.
FIMD integrates the key aspects of the human disease that an animal model must simulate. Naturally, no model is expected to mimic the human condition fully. However, understanding which features an animal model can and which it cannot replicate allows researchers to select optimal models for their research question. More importantly, it puts the results from animal studies into the broader context of human biology, potentially preventing the advancement of clinical trials based on data unlikely to translate.
3. Systematic Review and Meta-Analysis
Systematic reviews and meta-analyses of animal studies were one of the earliest tools to expose the status of the field [33]. Nonetheless, their application to compare animal models of disease is relatively recent. In FIMD’s pilot study, the two models of type 2 diabetes (the ZDF rat and db/db mouse) only presented slight differences in the general score. Since FIMD does not compare models quantitatively, we conducted a systematic review and meta-analysis to compare the effect of glucose-lowering agents approved for human use on the HbA1c [34]. We chose HbA1c as the outcome owing to its clinical relevance in type 2 diabetes drug development [35][36].
The results largely confirmed FIMD’s pilot study results—both models responded similarly to drugs, irrespective of the mechanism of action. The only exception was exenatide, which led to higher reductions in HbA1c in the ZDF rat. Both models predicted the direction of effect in humans for drugs with enough studies. Moreover, the quality assessment showed that animal studies are poorly reported: no study mentioned blinding at any level, and less than half reported randomisation. In this context, the risk of bias could not be reliably assessed.
The development of systematic reviews and meta-analyses to combine animal and human data offers an unprecedented opportunity to investigate the value of animal models of disease further. Nevertheless, translational meta-analyses are still uncommon [37]. A prospect for another application of systematic reviews and meta-analyses lies in comparing drug effect sizes in animals and humans directly. By calculating the degree of overlap of preclinical and clinical data, animal models could be ranked according to the extent they can predict effect sizes across different mechanisms of action and drug classes. The calculation of this ‘translational coefficient’ would include effective and ineffective drugs. Using human effect sizes as the denominator, a translational coefficient higher than 1 would indicate an overestimation of treatment effect, while a coefficient lower than 1, an underestimation. The systematisation of translational coefficients would lead to ‘translational tables’, giving additional insight on models’ translatability. These translational tables, allied with more qualitative approaches, such as FIMD, could form the basis for evidence-based animal model selection in the future.
Indeed, such a strategy is not without shortcomings. Owing to significant differences in the methodology of preclinical and clinical studies, such comparisons may present unusually large confidence intervals, complicating their interpretation. In addition, preclinical studies would need to match the standards of clinical research to a higher degree, including the use of more relevant endpoints that can be compared. Considerations on other design (e.g., dosing, route of administration), statistical (e.g., sample size, measures of spread) and biological matters (species differences) will be essential to develop a scientifically sound approach.
4. Can Animal Models Predict Human Pharmacologically Active Ranges? A First Glance into the Investigator’s Brochure
The decision to proceed to first-in-human trials is mostly based on the Investigator’s Brochure (IB), a document required by the Good Clinical Practice (GCP) guidelines [38]. The IB compiles all the necessary preclinical and clinical information for ethics committees and investigators to evaluate a drug’s suitability to be tested in humans. The preclinical efficacy and safety data in the IB are, thus, the basis for the risk-benefit analysis at this stage. Therefore, it is paramount that these experiments are performed to the highest standards to safeguard healthy volunteers and patients.
However, the results from Wieschoswki and colleagues show a different scenario [26]. They analysed 109 IBs presented for ethics review of three German institutional review boards. The results showed that the vast majority of preclinical efficacy studies did not report measures to prevent bias, such as blinding or randomisation. Furthermore, these preclinical studies were hardly ever published in peer-reviewed journals and were overwhelmingly positive—only 6% of the studies reported no significant effect. The authors concluded IBs do not provide enough high-quality data to allow a proper risk-benefit evaluation of investigational products for first-in-human studies during the ethics review.
In an ongoing study to investigate the predictivity of the preclinical data provided in the Ibs, we are evaluating whether animal models can predict pharmacologically active ranges in humans. Since pharmacokinetic (PK) and pharmacodynamic (PD) data are often scattered throughout the IB across several species, doses and experiments, integrating it can be challenging. We are using the IB-derisk, a tool developed by the Centre for Human Drug Research (CHDR), to facilitate this analysis [39]. The IB-derisk consists of a colour-coded excel sheet or web application (www.ib-derisk.org) in which PK and PD data can be inputted. It allows the extra- and interpolation of missing PK parameters across animal experiments, facilitating dose selection in first-in-human trials. The IB-derisk yields yet another method to discriminate between animal models of disease. With sufficient data, the drug PK and PD from preclinical studies of a single model and clinical trials of correspondent drugs can be compared. This analysis, when combined with PK/PD modelling, can serve as a tool to select the most relevant animal model based on the mechanism of action and model characteristics. Preliminary (and unpublished) results suggest that animal models can often predict human pharmacologically active ranges. How the investigated pharmacokinetic parameters relate to indication, safety, and efficacy is still unclear.
To build on Wieschowski’s results, we have been collecting data on the internal validity and reporting quality of animal experiments. Our initial analysis indicates the included IBs also suffer from the same pitfalls identified by Wieschowski, suggesting that such problems are likely to be widespread. In addition, only a few IBs justified their choice of model(s) of disease, and none compared their model(s) to other options to better understand their pros and cons. This missing information is crucial to allow for risk–benefit analysis during the ethics review process.
5. Can Animal Models Predict Human Pharmacologically Active Ranges? A First Glance into the Investigator’s Brochure
By applying FIMD, systematic reviews and meta-analysis, and the IB-derisk, researchers can identify more predictive disease models, potentially preventing clinical trials starting based on unreliable data. Ethics committees have a unique opportunity to incentivise higher standards since an unfavourable assessment can prevent poorly designed experiments from even starting. The request of a more detailed translational rationale for each model choice (e.g. by requiring models are evaluated with FIMD), as well as the enforcement of reporting guidelines, can act as gatekeepers for flawed study designs and improve the risk-benefit analysis significantly.
Funders can require the use of systematic reviews and meta-analyses and a thorough assessment of the translational relevance of selected animal models (e.g. FIMD). Journal editors and reviewers must actively enforce reporting guidelines for ongoing submissions as endorsing them does not improve compliance. Regulatory agencies can shape the drug development landscape significantly by, for instance, updating the IB guidelines by requiring a more extensive translational rationale for each animal model employed. Scientific advice can be used as a platform to discuss translational considerations early in development.
Larger companies can perform a thorough assessment of preclinical data of internal and external assets using FIMD, systematic review and meta-analysis, and the IB-derisk. At the same time, small and medium enterprises can provide data in these formats to support their development plan. Ultimately, the selection of more predictive disease models will lead to more successful clinical trials, increasing the benefit and reducing the risks to patients, and lower development costs.
A positive side-effect of these strategies is the increased scrutiny of design and animal model choices. Instead of a status-quo based on tradition and replication of poor practices, we can move forward to an inquisitive and evidence-based modus operandi. This change of culture is sorely needed in both academic and industrial institutions. A shift toward a stricter approach – more similar to clinical trials, from beginning to end, is warranted. The harmonisation of requirements across stakeholders will be crucial for a successful change of mindset.
In the foreseeable future, animal research is unlikely to be eliminated. The criticism of the present state of affairs of preclinical research is indeed justifiable. Our efforts must be focused on improving the robustness of animal data generated now. We already have tools available to address most, if not all, internal and external validity concerns. Only a thorough assessment of higher-quality animal data will determine whether animal research is still a valid paradigm in drug development. By promoting and adopting high-quality study design and reporting as well as a thorough assessment of the translatability of drug efficacy of animal models of disease, we will have robust data to improve the current animal research paradigm.
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715, doi:10.1038/nrd1470.
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286, doi:10.1093/biostatistics/kxx069.
- Pammolli, F.; Righetto, L.; Abrignani, S.; Pani, L.; Pelicci, P.G.; Rabosio, E. The endless frontier? The recent increase of R&D productivity in pharmaceuticals. J. Transl. Med. 2020, 18, 162, doi:10.1186/s12967-020-02313-z.
- Van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, e1000245, doi:10.1371/journal.pmed.1000245.
- Schulz, J.B.; Cookson, M.R.; Hausmann, L. The impact of fraudulent and irreproducible data to the translational research crisis—Solutions and implementation. J. Neurochem. 2016, 139, 253–270, doi:10.1111/jnc.13844.
- Ioannidis, J.P.A. Acknowledging and overcoming nonreproducibility in basic and preclinical research. JAMA 2017, 317, 1019, doi:10.1001/jama.2017.0549.
- Vogt, L.; Reichlin, T.S.; Nathues, C.; Würbel, H. Authorization of animal experiments is based on confidence rather than evidence of scientific rigor. PLoS Biol. 2016, 14, e2000598, doi:10.1371/journal.pbio.2000598.
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533, doi:10.1038/483531a.
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712–712, doi:10.1038/nrd3439-c1.
- Perrin, S. Preclinical research: Make mouse studies work. Nature 2014, 507, 423–425, doi:10.1038/507423a.
- Pound, P.; Ritskes-Hoitinga, M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med. 2018, 16, 304, doi:10.1186/s12967-018-1678-1.
- Bebarta, V.; Luyten, D.; Heard, K. Emergency medicine animal research: Does use of randomization and blinding affect the results? Acad. Emerg. Med. 2003, 10, 684–687, doi:10.1111/j.1553-2712.2003.tb00056.x.
- Schmidt‐Pogoda, A.; Bonberg, N.; Koecke, M.H.M.; Strecker, J.; Wellmann, J.; Bruckmann, N.; Beuker, C.; Schäbitz, W.; Meuth, S.G.; Wiendl, H.; et al. Why most acute stroke studies are positive in animals but not in patients: A systematic comparison of preclinical, early phase, and phase 3 clinical trials of neuroprotective agents. Ann. Neurol. 2020, 87, 40–51, doi:10.1002/ana.25643.
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412, doi:10.1371/journal.pbio.1000412.
- Smith, A.J.; Clutton, R.E.; Lilley, E.; Hansen, K.E.A.; Brattelid, T. PREPARE: Guidelines for planning animal research and testing. Lab. Anim. 2018, 52, 135–141, doi:10.1177/0023677217724823.
- Osborne, N.; Avey, M.T.; Anestidou, L.; Ritskes‐Hoitinga, M.; Griffin, G. Improving animal research reporting standards: HARRP, the first step of a unified approach by ICLAS to improve animal research reporting standards worldwide. EMBO Rep. 2018, 19, doi:10.15252/embr.201846069.
- Baker, D.; Lidster, K.; Sottomayor, A.; Amor, S. Two years later: Journals are not yet enforcing the ARRIVE guidelines on reporting standards for pre-clinical animal studies. PLoS Biol. 2014, 12, e1001756, doi:10.1371/journal.pbio.1001756.
- Henderson, V.C.; Kimmelman, J.; Fergusson, D.; Grimshaw, J.M.; Hackam, D.G. Threats to validity in the design and conduct of preclinical efficacy studies: A systematic review of guidelines for in vivo animal experiments. PLoS Med. 2013, 10, e1001489, doi:10.1371/journal.pmed.1001489.
- Hackam, D.G. Translating animal research into clinical benefit. BMJ 2007, 334, 163–164, doi:10.1136/bmj.39104.362951.80.
- Sams-Dodd, F. Strategies to optimize the validity of disease models in the drug discovery process. Drug Discov. Today 2006, 11, 355–363, doi:10.1016/j.drudis.2006.02.005.
- Ferreira, S.G.; Veening-Griffioen, D.H.; Boon, W.P.C.; Moors, E.H.M.; Gispen-de Wied, C.C.; Schellekens, H.; van Meer, P.J.K. A standardised framework to identify optimal animal models for efficacy assessment in drug development. PLoS ONE 2019, 14, e0218014, doi:10.1371/journal.pone.0218014.
- Ferreira, G.S.; Veening-Griffioen, D.H.; Boon, W.P.C.; Moors, E.H.M.; Gispen-de Wied, C.C.; Schellekens, H.; van Meer, P.J.K. Correction: A standardised framework to identify optimal animal models for efficacy assessment in drug development. PLoS ONE 2019, 14, e0220325, doi:10.1371/journal.pone.0220325.
- Macleod, M.R.; Lawson McLean, A.; Kyriakopoulou, A.; Serghiou, S.; de Wilde, A.; Sherratt, N.; Hirst, T.; Hemblade, R.; Bahor, Z.; Nunes-Fonseca, C.; et al. Risk of bias in reports of in vivo research: A focus for improvement. PLoS Biol. 2015, 13, e1002273, doi:10.1371/journal.pbio.1002273.
- Langhof, H.; Chin, W.W.L.; Wieschowski, S.; Federico, C.; Kimmelman, J.; Strech, D. Preclinical efficacy in therapeutic area guidelines from the U.S. Food and Drug Administration and the European Medicines Agency: A cross-sectional study. Br. J. Pharmacol. 2018, 175, 4229–4238, doi:10.1111/bph.14485.
- Varga, O.E.; Hansen, A.K.; Sandøe, P.; Olsson, I.A.S. Validating animal models for preclinical research: A scientific and ethical discussion. Altern. Lab. Anim. 2010, 38, 245–248, doi:10.1177/026119291003800309.
- Wieschowski, S.; Chin, W.W.L.; Federico, C.; Sievers, S.; Kimmelman, J.; Strech, D. Preclinical efficacy studies in investigator brochures: Do they enable risk–benefit assessment? PLoS Biol. 2018, 16, e2004879, doi:10.1371/journal.pbio.2004879.
- McKinney, W.T. Animal model of depression: I. Review of evidence: Implications for research. Arch. Gen. Psychiatry 1969, 21, 240, doi:10.1001/archpsyc.1969.01740200112015.
- Willner, P. The validity of animal models of depression. Psychopharmacology 1984, 83, 1–16, doi:10.1007/BF00427414.
- Denayer, T.; Stöhr, T.; Roy, M.V. Animal models in translational medicine: Validation and prediction. Eur. J. Mol. Clin. Med. 2014, 2, 5, doi:10.1016/j.nhtm.2014.08.001.
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.M.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43, doi:10.1186/1471-2288-14-43.
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213, doi:10.1242/dmm.018424.
- Yu, X.; Bao, B.; Echigoya, Y.; Yokota, T. Dystrophin-deficient large animal models: Translational research and exon skipping. Am. J. Transl. Res. 2015, 7, 1314–1331.
- Pound, P.; Ebrahim, S.; Sandercock, P.; Bracken, M.B.; Roberts, I. Where is the evidence that animal research benefits humans? BMJ 2004, 328, 514–517, doi:10.1136/bmj.328.7438.514.
- Ferreira, G.S.; Veening-Griffioen, D.H.; Boon, W.P.C.; Hooijmans, C.R.; Moors, E.H.M.; Schellekens, H.; van Meer, P.J.K. Comparison of drug efficacy in two animal models of type 2 diabetes: A systematic review and meta-analysis. Eur. J. Pharmacol. 2020, 879, 173153, doi:10.1016/j.ejphar.2020.173153.
- FDA. Guidance for Industry Diabetes Mellitus: Developing Drugs and Therapeutic Biologics for Treatment and Prevention; FDA: Washington, DC, USA, 2008.
- EMA. Guideline on Clinical Investigation of Medicinal Products in 5 the Treatment or Prevention of Diabetes Mellitus; FDA: Washington, DC, USA, 2018.
- Leenaars, C.H.C.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; De Vries, R.B.M.; Meijboom, F.L.B. Animal to human translation: A systematic scoping review of reported concordance rates. J. Transl. Med. 2019, 17, 223, doi:10.1186/s12967-019-1976-2.
- European Medicines Agency. ICH E6 (R2) Good Clinical Practice—Step 5; European Medicines Agency: London, UK, 2016.
- Van Gerven, J.; Cohen, A. Integrating data from the investigational medicinal product dossier/investigator’s brochure. A new tool for translational integration of preclinical effects. Br. J. Clin. Pharmacol. 2018, 84, 1457–1466, doi:10.1111/bcp.13529.


