 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Janusz Blasiak | + 2480 word(s) | 2480 | 2020-07-24 04:18:07 | | | |
| 2 | Bruce Ren | -34 word(s) | 2446 | 2020-07-29 10:56:54 | | | | |
| 3 | Bruce Ren | -34 word(s) | 2446 | 2020-07-29 10:57:27 | | | | |
| 4 | Bruce Ren | Meta information modification | 2446 | 2020-07-30 02:29:58 | | |
Video Upload Options
Zinc supplementation is reported to slow down the progression of age-related macular degeneration (AMD), but there is no general consensus on the beneficiary effect on zinc in AMD. As zinc can stimulate autophagy that is declined in AMD, it is rational to assume that it can slow down its progression. As melanosomes are the main reservoir of zinc in the retina, zinc may decrease the number of lipofuscin granules that are substrates for autophagy. The triad zinc–autophagy–AMD could explain some controversies associated with population studies on zinc supplementation in AMD as the effect of zinc on AMD may be modulated by genetic background. This aspect was not determined in many studies regarding zinc in AMD. Zinc deficiency induces several events associated with AMD pathogenesis, including increased oxidative stress, lipid peroxidation and the resulting lipofuscinogenesis. The latter requires autophagy, which is impaired. This is a vicious cycle-like reaction that may contribute to AMD progression. Promising results with zinc deficiency and supplementation in AMD patients and animal models, as well as emerging evidence of the importance of autophagy in AMD, are the rationale for future research on the role of autophagy in the role of zinc supplementation in AMD.
1. Introduction
Age-related macular degeneration (AMD) is the main cause of legal blindness in the elderly in the Western World, and zinc supplementation is reported in several studies to have a beneficial effect for AMD patients. However, some studies report no effect and others, rare—even an adverse influence. It is not completely clear how zinc deficiency may influence AMD pathogenesis. It is important to address this problem as the European Food Safety Authority Panel reported that a cause and effect relationship between the dietary intake of zinc and the maintenance of normal vision had been satisfactorily established (cited in [1]).
Autophagy, an evolutionary conserved process that serves the cell by removing and recycling damaged or no longer used cellular components, declines with age and its impairment is associated with several human age-related diseases, including AMD (reviewed in [2]). However, the role of autophagy in AMD pathogenesis, similar to its general role, raises many unanswered questions (reviewed in [3]).
Zinc may modulate, in many cases stimulate, autophagy, although not all details of this modulation are certain (reviewed in [4]). On the other hand, impaired autophagy may change cellular zinc homeostasis.
In summary, we can consider mutual relationships between AMD pathogenesis and zinc deficiency, impaired autophagy and zinc deficiency, as well as AMD pathogenesis and impaired autophagy. Therefore, the ternary relationship between zinc, autophagy, and AMD is logical, but it has not been shown so far. In this review, we present and update information on mutual relationships within the triad zinc–autophagy–AMD and provide arguments that at least a part of the protective effect of zinc against AMD can be attributed to the modulation of autophagy by zinc.
2. Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is a complex eye disease that is the main cause of legal blindness in the elderly in industrialized countries. Usually, two phases of the disease are distinguished: early and late (advanced), although this categorization fails in some cases. AMD is strongly correlated with age as the prevalence of its early form in Europe increased from 3.5% in subjects aged 55–59 years to 17.6% in individuals aged greater or equal to 85 years; for late AMD, these ratios were 0.1% and 9.8%, respectively [5]. The occurrence of AMD in the United States (US) is projected to increase to 22 million by the year 2050, while the global prevalence is anticipated to increase to 288 million by the year 2040 [5][6]. The dependence on age was strengthened by the discovery of an increased level of amyloid-beta in the aging retina and AMD retinas [7]. Due to this fact, AMD is sometimes described as “dementia of the eye” or “Alzheimer’s disease in the eye” [8].
AMD affects the macula—a small structure in the center of the retina that is responsible for sharp central and color vision. AMD causes vision loss due to non-functional or dead photoreceptors and underlying retinal pigment epithelium (RPE) cells [7] (Figure 1). The RPE contains polarized epithelial cells contacting photoreceptor outer segments (POSs), while their other part contacts Bruch’s membrane (BM) between the RPE and choriocapillaris. Phenotypically, advanced AMD can be divided into two basic forms: dry (atropic, non-exudative) and wet (exudative). In its dry form, the disease is initiated by the formation of drusen and pigment mottling associated with dysfunctions in RPE cells. Dry AMD may progress to geographic atrophy (GA) associated with the loss of RPE and photoreceptors. In wet AMD, RPE remains integral but initiates the production of angiogenic agents, including vascular endothelial growth factor (VEGF) that stimulate the formation of new blood vessels from underlying choriocapillaris. This process is termed choroidal neovascularization (CNV) and may lead to photoreceptors’ death. Dysfunction of choriocapillaris is often observed in both AMD types.
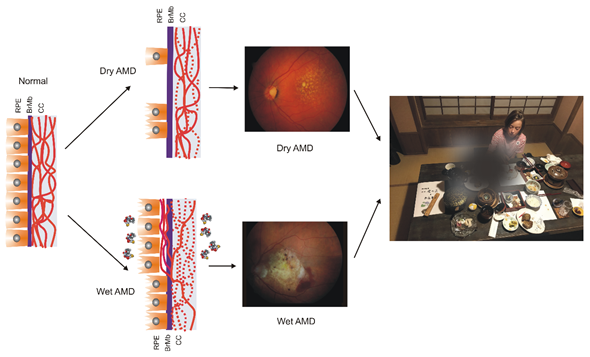
Figure 1. Age-related macular degeneration (AMD). The normal retina contains photoreceptors, not shown here, that interact with the retinal pigment epithelium (RPE)/Bruch’s membrane/choriocapillaris (CC) complex, which is supported by the large choroidal blood vessels (not presented). In dry AMD, some RPE cells are damaged, and some are lost when the disease progresses. Some CCs are also lost (broken lines) in dry AMD, but in the wet form of the disease, they are lost almost completely. In wet AMD, RPE cells release several factors (multicolor small objects) that support angiogenesis, including vascular endothelial growth factor, which leads to choroidal neovascularization. Fundus color images of dry and wet AMD. Light orange spots in the retina termed drusen can be observed in both dry and wet AMD, whereas wet AMD is featured by hemorrhages and edema. AMD can ruin the central vision of affected individuals.
Aging is the most serious risk factors associated with AMD. Familial history and genetic variability in several loci, mostly associated with the complement system, are reported to increase AMD risk [9]. Many environmental and lifestyle influences are presented as AMD risk factors, including smoking, fat-rich diet and obesity, UV and blue light exposure, female sex, light color of the iris, associated cardiovascular disease, high blood pressure, but the causality of these factors is problematic. Oxidative stress is frequently mentioned as an AMD risk factor, but it is associated with almost all the previously mentioned factors, including aging. Moreover, the retina has the highest metabolic rate among all tissues of the human body, and it produces reactive oxygen species (ROS) even in its normal functioning, and this production increases with AMD-associated changes. It was shown that RPE obtained from AMD donors displayed increased levels of ROS and a higher susceptibility to oxidative stress than RPE of non-AMD donors [10]. Therefore, it can be concluded that oxidative stress is associated with AMD, but its role in the pathogenesis of this disease requires further research to show its source and consequences.
3. Autophagy in Age-Related Macular Degeneration
Autophagy is an essential cellular process in which cellular components that are damaged, dysfunctional, or no longer needed (autophagic cargo) are degraded in the lysosome and recycled. This process can be categorized into macro autophagy (hereafter termed autophagy), micro autophagy, and chaperone-mediated autophagy.
Autophagy has several clearly distinguished steps (Figure 2). In the first stage, autophagy is initiated, then nucleation of the double membrane and its elongation (phagophore formation) occur, followed by the encapsulation of the cargo and fusion with completely enclosed phagophore (autophagosome) takes place. Autophagosome then fuses with lysosome to form autolysosome, in which the cargo is degraded (reviewed in [11]). Many autophagy-related proteins (ATGs) are involved in autophagy; some of them are evolutionary conserved. Mechanistic target of rapamycin (mTOR), a nutrient sensor, is a major upstream regulator of autophagy along with AMP-activated kinase that phosphorylates Unc−51-like kinase 1 (ULK1). Microtubule-associated protein light chain 3 (LC3) and γ-aminobutyric acid receptor-associated proteins (GABARAPs) form a protein family of a key significance in autophagy. The LC3/GABARAP family proteins attach to the auto lysosomal membrane and interact with various autophagy receptors, including p62/SQSTM1 (sequestesome 1).
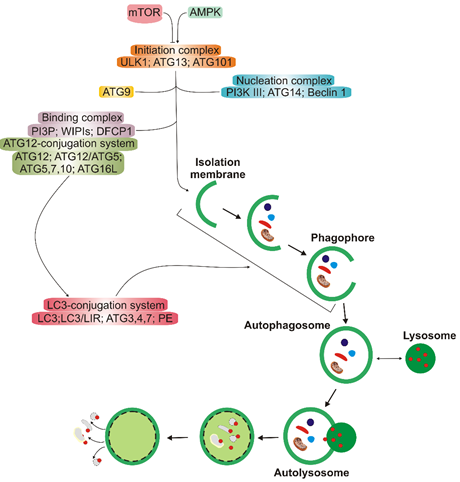
Figure 2. A simplified scheme of macro autophagy (further: autophagy). Mechanistic targets of rapamycin (mTOR) and AMP-activated kinase (AMPK) are the primary autophagy inhibitor and inducer, respectively. On autophagy initiation, the cytoplasmic material to be degraded (cargo) is gradually encapsulated by double membranes: phagophores to encapsulated vesicles (autophagosomes), which then fuse with lysosomes to form autolysosomes, where cargo is degraded and then released into cytoplasm to be used again (recycling). Autophagy is regulated by many proteins, the autophagy-related proteins (ATGs), and their complexes. The initiation process is led by the Unc − 51-like kinase 1 (ULK1) complex, the formation of the double membrane, provided by ATG − 9-containing vesicles, and autophagosome—by the class III PI3K (phosphatidylinositol 3-kinase) nucleation complex and the phosphatidylinositol 3-phosphate (PI3P)-binding complex, which includes ATG12, Beclin 1, and the microtubule-associated protein light chain 3/γ-aminobutyric acid receptor-associated proteins (LC3/GABARAPs), represented here only by LC3. ATG12 attaches to ATG5, which is then associated with ATG16L1. This complex stimulates the cleavage of LC3 by ATG4 to form LC3-I, which is then conjugated with phosphatidylethanolamine (PE) to form LC3-II. On the incorporation into autophagosomal membranes, LC3 interacts with cargo receptors, which have LC3-interacting motifs (LIRs).
Autophagy is not only a stress-related process but it occurs at the basal level in normal conditions to remove byproducts of normal metabolism and is involved in many life processes. As Daniel Klionsky said, “Autophagy participates in, well, just about everything” [12].
As mentioned, oxidative stress in the retina associates AMD. Reactive oxygen and nitrogen species damage cellular biomolecules, including proteins that can be misfolded by oxidation and glutathione conjugation [13]. This kind of protein damage evokes the unfolded protein response (UPR) and can be repaired by molecular chaperones. However, if this system is not efficient, soluble proteins are ubiquitinated and targeted to degradation in the proteasome (reviewed in[14]). Furthermore, proteasomal degradation may fail or dysfunction in aging and neurodegeneration, which may lead to the accumulation of oxidized and ubiquitinated proteins (reviewed in [15]). The ubiquitination attracts autophagy receptors, such as p62/SQSTM1 and LC3, that link these cellular waste to autophagy. When autophagy fails in RPE cells, this may lead to the accumulation of lipofuscin and activation of the NLRP3 (NLR family pyrin domain-containing 3) inflammasome, eventually resulting in drusen formation and low-grade chronic inflammation in the retina, accelerating the aging process (reviewed in [16]). However, Kosmidou et al. pointed to the need for re-interpretation of published results reporting NLRP3 expression and upregulation in human and human-derived RPE cells and addressed the role that the inflammasome plays in AMD pathogenesis [17].
Non-exudative (dry) AMD seems to be of special interest from the perspective of autophagy as this form of the disease is associated with the accumulation of lipofuscin on the RPE. Lipofuscin contains lysosomal insoluble pigment granules that remain after lysosomal digestion [18]. Impaired autophagy favors lipofuscin formation, and it has been observed that rapamycin, an autophagy inducer, decreased lipofuscin accumulation in RPE cells [19]. Rapamycin also ameliorated cellular damage in the RPE induced by A2E, a lipofuscin fluorophore [20].
As autophagy contributes to the maintenance of cellular homeostasis, its high level is observed in stress conditions [21]. However, such an increase in autophagy is often observed along with cellular death, which led to the conclusion that besides pro-life functions, autophagy may act as a cellular death executioner (pro-death functions, reviewed in [22]). The mechanism of the regulation of these two pathways of autophagy is not completely clear, and it is not easy to relate them to AMD pathogenesis. As suggested by Mitter et al., autophagy might be dysregulated in two ways in AMD [19]. They observed that acute oxidative stress might increase autophagy in RPE cells, while chronic oxidative stress was associated with reduced autophagy. Therefore, when oxidative stress was induced in RPE cells, it might induce autophagy that acted in two phases—the initial increase followed by a decrease. In conclusion, Mitter et al. postulated that autophagy was important for the protection of the RPE against oxidative stress, and lipofuscin accumulation and impaired autophagy might aggravate oxidative stress and in this way play an important role in AMD pathogenesis.
4. Conclusions and Perspectives
Several studies on AMD patients and animal models, as well as cell cultures, suggest that zinc may play a positive role in AMD pathogenesis. The exact mechanism of the role of zinc deficiency plays in AMD is not known. Due to mutual relationship between zinc and AMD, zinc and autophagy as well as autophagy and AMD, we suggest that the effect of zinc in AMD can be determined by autophagy and this may be the reason that some studies do not confirm beneficiary effects of zinc in AMD, as was shown in AREDS/AREDS2, and other studies report a negative consequence of zinc supplementation for AMD patients. However, these “negative” studies are rare, and their research design is questionable, and that is why they were not considered in the context of autophagy in AMD in this manuscript [23][24][25][26].
As shown in the Rotterdam Eye Study, the effect on zinc on the vision might depend on individual genetic background [27]. In the post-genomic era, it is tempting to relate the reaction of zinc to an individual’s complete genetic profile, but it has little sense, if any. Who could elaborate the algorithm for the use of AREDS-like formulation to prevent AMD or slow down its progression and decrease consequences? Instead, a dedicated microarray could be projected with genes that are important for AMD pathogenesis and zinc metabolism. Of course, the main problem would be with the selection of AMD pathogenesis genes. In the context of the research performed so far, AMD-susceptibility genes, genes for zinc metabolism, and genes of autophagy could be included.
Altogether, the influence of zinc on AMD patients is a complex issue as many pathways can be involved, including autophagy. Moreover, autophagy in AMD is also a complicated subject with no unequivocal determination of all mechanisms lying beyond observed and expected effects. As mentioned, it seems that the transport of melanosomes and lipofuscin accumulation may be an important connection in zinc deficiency, autophagy, and AMD. Recently, Xia et al. showed that missense mutations in the UBE3D (Ubiquitin Protein Ligase E3D) gene, which was identified to be associated with wet AMD, were linked with several adverse effects in the retina, including delayed retrograde melanosome transport, increased deposition of pigment granules, and impaired autophagy in zebrafish [28]. As shown by Julien et al., the metal ion concentration of RPE melanosomes is regulated by zinc, and reduced metal-binding activity of melanosomes is associated with degenerative processes in the retina [29]. Therefore, melanosomes and their properties,
References
- Gilbert, R.; Peto, T.; Lengyel, I.; Emri, E. Zinc Nutrition and Inflammation in the Aging Retina. Mol. Nutr. Food Res. 2019, 63, e1801049, doi:10.1002/mnfr.201801049.
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593, doi:10.1038/s41580-018-0033-y.
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984, doi:10.4161/auto.24546.
- Liuzzi, J.P.; Guo, L.; Yoo, C.; Stewart, T.S. Zinc and autophagy. Biomet. Int. J. Role Met. Ions Biol. Biochem. Med. 2014, 27, 1087–1096, doi:10.1007/s10534-014-9773-0.
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763, doi:10.1016/j.ophtha.2017.05.035.
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116, doi:10.1016/s2214-109x(13)70145-1.
- Ratnayaka, J.A.; Serpell, L.C.; Lotery, A.J. Dementia of the eye: The role of amyloid beta in retinal degeneration. Eye (London) 2015, 29, 1013–1026, doi:10.1038/eye.2015.100.
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J. Alzheimers Dis. JAD 2011, 24, 615–631, doi:10.3233/jad-2011-101908.
- Yoneyama, S.; Sakurada, Y.; Kikushima, W.; Sugiyama, A.; Matsubara, M.; Fukuda, Y.; Tanabe, N.; Parikh, R.; Mabuchi, F.; Kashiwagi, K.; et al. Genetic factors associated with response to as-needed aflibercept therapy for typical neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Sci. Rep. 2020, 10, 7188, doi:10.1038/s41598-020-64301-z.
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537, doi:10.1038/cddis.2016.453.
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis Nat. Rev. Mol. Cell Biol. 2020, 10.1038/s41580-020-0241-0, doi:10.1038/s41580-020-0241-0.
- Klionsky, D.J. Autophagy participates in, well, just about everything. Cell Death Differ. 2020, 27, 831–832, doi:10.1038/s41418-020-0511-6.
- Nakamura, T.; Lipton, S.A. Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell. Mol. Life Sci. CMLS 2007, 64, 1609–1620, doi:10.1007/s00018-007-6525-0.
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, doi:10.3390/cells8010040.
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712, doi:10.1038/s41580-018-0040-z.
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. CMLS 2016, 73, 1765–1786, doi:10.1007/s00018-016-2147-8.
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration. Sci. Rep. 2018, 8, 461, doi:10.1038/s41598-017-17634-1.
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619, doi:10.1016/s0891-5849(02)00959-0.
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005, doi:10.4161/auto.36184.
- Zhang, J.; Bai, Y.; Huang, L.; Qi, Y.; Zhang, Q.; Li, S.; Wu, Y.; Li, X. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: Implications for age-related macular degeneration. Cell Death Dis. 2015, 6, e1972, doi:10.1038/cddis.2015.330.
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. 2019, 94, 503–516, doi:10.1111/brv.12464.
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42, doi:10.1038/icb.2014.85.
- Cho, E.; Stampfer, M.J.; Seddon, J.M.; Hung, S.; Spiegelman, D.; Rimm, E.B.; Willett, W.C.; Hankinson, S.E. Prospective study of zinc intake and the risk of age-related macular degeneration. Ann. Epidemiol. 2001, 11, 328–336, doi:10.1016/s1047-2797(01)00217-4.
- Chong, E.W.; Wong, T.Y.; Kreis, A.J.; Simpson, J.A.; Guymer, R.H. Dietary antioxidants and primary prevention of age related macular degeneration: Systematic review and meta-analysis. BMJ (Clin. Res. Ed.) 2007, 335, 755, doi:10.1136/bmj.39350.500428.47.
- Evans, J.R. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Systematic Rev. 2006, 10.1002/14651858.CD000254.pub2, Cd000254, doi:10.1002/14651858.CD000254.pub2.
- Evans, J.R.; Henshaw, K. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Systematic Rev. 2008, 10.1002/14651858.CD000253.pub2, Cd000253, doi:10.1002/14651858.CD000253.pub2.
- Ikram, M.A.; Brusselle, G.G.O.; Murad, S.D.; van Duijn, C.M.; Franco, O.H.; Goedegebure, A.; Klaver, C.C.W.; Nijsten, T.E.C.; Peeters, R.P.; Stricker, B.H.; et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 2017, 32, 807–850, doi:10.1007/s10654-017-0321-4.
- Xia, H.; Zhang, Q.; Shen, Y.; Bai, Y.; Ma, X.; Zhang, B.; Qi, Y.; Zhang, J.; Hu, Q.; Du, W.; et al. ube3d, a New Gene Associated with Age-Related Macular Degeneration, Induces Functional Changes in Both In Vivo and In Vitro Studies. Mol. Ther. Nucleic Acids 2020, 20, 217–230, doi:10.1016/j.omtn.2020.02.010.
- Julien, S.; Biesemeier, A.; Kokkinou, D.; Eibl, O.; Schraermeyer, U. Zinc deficiency leads to lipofuscin accumulation in the retinal pigment epithelium of pigmented rats. PLoS ONE 2011, 6, e29245, doi:10.1371/journal.pone.0029245.


