 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fausto Sanchez-Munoz | + 1738 word(s) | 1738 | 2020-07-13 06:09:01 | | | |
| 2 | Nora Tang | -1 word(s) | 1737 | 2020-07-20 11:03:45 | | | | |
| 3 | Nora Tang | Meta information modification | 1737 | 2020-07-21 07:53:59 | | |
Video Upload Options
Pulmonary arterial hypertension (PAH) is a severe disease characterized by the loss and obstructive remodeling of the pulmonary arterial wall, causing a rise in pulmonary arterial pressure and pulmonary vascular resistance, which is responsible for right heart failure, functional decline, and death. The main molecular pathways involved in drugs are available for the treatment PAH involve nitric oxide, endothelin 1 and prostacyclin. Although, this condition continues to be life-threatening, and its long-term treatment is expensive.
1. Definition
Pulmonary arterial hypertension (PAH) is a life-threatening disease characterized by the obstruction, loss, and remodeling of the pulmonary arteries, which increases pulmonary arterial pressure (PAP) and pulmonary vascular resistance (PVR). Hemodynamically, PAH is defined by a mean pulmonary artery pressure (mPAP) of ≥20 mm Hg, a pulmonary arterial wedge pressure (PAWP) of ≥15 mm Hg, and a pulmonary vascular resistance (PVR) of ≥three Wood Units (WU), as established at the sixth World Symposium on Pulmonary hypertension (PH) in Nice 2018 [1].
2. Introduction
PAH prevalence has increased over time. Worldwide, the latest records show an incidence of 2.0 to 7.6 cases per million adults per year and a prevalence of 11 to 26 cases per million adults. The incidence is known to be four times higher among women, but survival is paradoxically worse in men [2][3]. PAH belongs to a group of severe clinical entities included in the current clinical classification of PH (Group 1). The other clinical scenarios include PH due to left heart disease (Group 2), PH due to lung disease or hypoxia (Group 3), PH due to chronic thromboembolic disease (Group 4), and PH due to unclear and/or multifactorial mechanisms (Group 5). In PAH, pulmonary vascular remodeling is characterized by an accumulation of vascular cells in the pulmonary arterial wall, such as changes in the pulmonary artery smooth muscle cells (PASMCs), endothelial cells, fibroblasts, myofibroblasts, and pericytes. This process can also involve the loss of [4]precapillary arteries and perivascular infiltration of inflammatory cells (macrophages, dendritic cells, mast cells, and B and T-lymphocytes) [4]. In general, pulmonary endothelial dysfunction contributes to pulmonary vascular remodeling in all groups of PAH [5][6]. Pulmonary endothelial cells (ECs) play a key role in the regulation of pulmonary vascular tone through the nitric oxide (NO), prostacyclin (PGI2), endothelin (ET), and serotonin (5-HT) pathways [7]. ECs are sensitive and respond to signals from extracellular environments where interactions with adjacent cells, circulating cells, and/or mediators help maintain a thrombosis-free surface. This property helps control inflammatory cell adhesion and assure normal angiogenesis, as well as the integrity of the vascular wall [8]. Conversely, the alteration or dysfunction of the pulmonary endothelium leads to an altered balance of the vasoconstriction and vasodilation mechanisms, as well as the acquisition of proinflammatory, prothrombotic, proproliferative, and antiapoptotic phenotypes [9].
The main mechanisms for the treatment of PAH are based on an imbalance of the NO, PGI2, and ET pathways [10]. Despite the existence of a wide variety of drugs for the treatment of PAH, it continues to be a life-threatening condition, and long-term treatment remains expensive. In this review, we discuss the current evidence of nutraceutical evidence in PAH models. Finally, we discuss the possibility to use combination therapy in PAH-specific drugs and nutraceuticals.
3. Molecular Pathways in the Treatment of PAH
The pathogenesis of pulmonary arterial hypertension is multifactorial and complex; therefore, the relevant treatments include several interventions, such as drugs, lifestyle and dietary changes, and surgery where necessary. Current knowledge of the pathophysiology of PAH has made it possible to distinguish three main signaling pathways that play a key role in the regulation of pulmonary vascular tone (nitric oxide, endothelin, and prostacyclin). Thus, available therapies are aimed at stimulating the local production of various vasoactive substances, including nitric oxide (NO) and prostacyclin, which contribute to the regulation of vascular tone in the pulmonary arteries. The drugs used in the treatment of PAH are used directly or indirectly to preserve, stimulate, or block the signaling pathways of vasoactive substances (Figure 1).
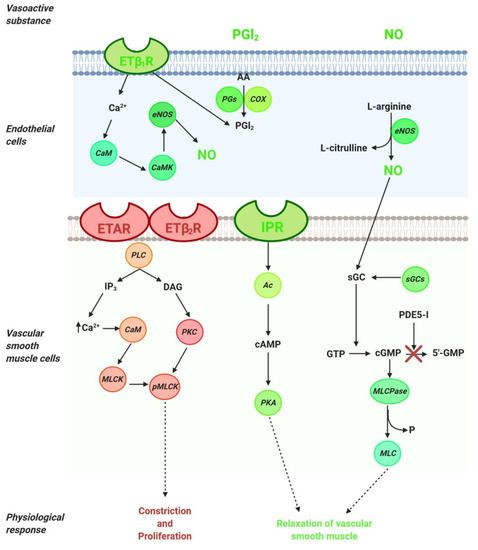
Figure 1. Signaling pathways in the regulation of pulmonary vascular tone. Arachidonic acid (AA); adenylyl cyclase (Ac); calmodulin (CaM); calmodulin kinase (CaMK);cyclooxygenase (COX); cyclic adenosine monophosphate (cAMP); cyclic guanosine monophosphate (cGMP); diacylglycerol (DAG); endothelial nitric oxide synthase (eNOS); endothelin 1 (ET-1); endothelin type A receptor (ETAR); endothelin type B1 receptor (ETB1R); endothelin type B2 receptor (ETB2R); guanosine monophosphate (GMP); inositol trisphosphate (IP3); I-prostanoid receptor (IPR); myosin light chain (MLC); myosin light chain kinase (MLCK); myosin light chain phosphatase (MLCPase); nitric oxide (NO); protein kinase A (PKA); prostaglandin synthase (PGs); protein kinase C (PKC); phosphodiesterase (PDE)-5 inhibitors (PDE-5i); phospholipase C (PLC); phosphorylated myosin light chain (pMLC); soluble guanylate cyclase stimulator (sGCs).
NO, the main endogenous vasodilator, is an easily diffusible gas that is synthesized in the endothelium from L-arginine by NO synthase. It diffuses vascular smooth muscle cells and mediates vascular relaxation through stimulation of soluble guanylate cyclase, thereby generating the second messenger cyclic guanosine monophosphate (cGMP). cGMP is metabolized by phosphodiesterase 5 (PDE-5) to GMP and inhibits NO-mediated vasodilation (Figure 1). Thus, in the treatment of PAH, PDE-5 inhibitors are considered a therapeutic option directed at the NO pathway. The drugs for PAH treatment include NO—cGMP enhancers (PDE-5 inhibitors), endothelin receptor antagonists and agonists of the prostacyclin pathways, and, recently, a soluble guanylate cyclase stimulator (sGCs). Additionally, riociguat works on a distinct molecular target in the same pathway as PDE-5 inhibitors.
3.1. NO Pathway
In the NO pathway, NO molecule binds to soluble guanylate cyclase (sGC) and leads to the production of cyclic guanosine monophosphate (cGMP), which activates myosin light chain phosphatase (MLCPase), thereby reducing the phosphorylation of myosin to induce arteriole vasodilation and inhibit cell proliferation (Figure 1). The phosphodiesterase-5 (PDE-5) enzyme is abundantly expressed in vascular smooth muscle cells (VSMCs) of the pulmonary vasculature and upregulated in the PAH in VSMCs and right ventricle cardiomyocytes. Moreover, the upregulation of PDE-5 leads to increased hydrolysis of cGMP [11][12]. PDE-5 inhibition prevents the degradation of cGMP to GMP (Figure 1), and sildenafil and tadalafil are two typical PDE-5 inhibitors that have demonstrated clinical benefits in PAH [13].
Riociguat is a new drug that acts independently of NO via the stimulation of sGC by increasing the generation of cGMP [14][15]. sGC is expressed in VSMCs of the pulmonary vasculature, platelets, the right ventricle, and other tissues. sGC binding to NO catalyzes the conversion of GTP to sGMP, thereby promoting beneficial effects, such as inhibiting smooth muscle proliferation leucocyte recruitment, inflammation, fibrosis, platelet aggregation, and vasodilation .
The combined use of both drugs (PDE-5 inhibitors and the stimulation of sGC) in patients with PAH is not recommended [16].
3.2. ET-1 Pathway
Endothelin (ET) is a peptide or vasoactive compound and is the most potent vasoconstrictor substance known [17]. ET-1 synthesis occurs when the inactive 39-amino-acid precursor pro-ET is cleaved by ET-converting enzymes to ET-1. ET-1 has two receptor subtypes (G protein-coupled receptors): the endothelin receptor A (ETAR), which is located on vascular smooth muscle cells, and endothelin receptor B (ETBR), which has two subtypes—ETB1R, located on endothelial cells, and ETB2R, located on vascular smooth muscle cells (Figure 1). The ETAR expressed in VSMCs and PAH contributes to contraction, proliferation, and proinflammatory effects [18][19]. On the other hand, ETB1R is expressed in vascular endothelial cells, where it acts to clear ET-1 and mediate the endothelial cell vasodilation through cytosolic Ca2+ binding to calmodulin to activate calmodulin kinase, which is responsible for phosphorylating endothelial nitric oxide synthase (NO) and synthesizing nitric oxide NO and PGI2 (Figure 1)[20][21] . In vascular smooth muscle cells, ET-1 binds to ETAR and ETB2R, thereby activating phospholipase C (PLC) to release inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 binds to its intracellular receptor, thus releasing calcium (Ca2+) into the cytosol to produce vasoconstriction and proliferative responses [22]. In PAH, ET-1 is upregulated and promotes PASMC proliferation [23][24].
In patients with PAH, ET-1 is overexpressed in the lung and plasma [24]. Bosentan, ambrisentan, and macitentan are endothelin receptor antagonists (ERAs) that have demonstrated to have beneficial effects in PAH [25][26][27].
ERAs were designed as either ETA-selective antagonists (ETA/ETB selectivity ratio >100) or dual ETA/ETB antagonists (equal selectivity), which suggests that dual antagonists have the potential to block ET-1-induced signaling more effectively than other ERAs [22]. However, the selective antagonists block ETA and vasoconstriction in VSMCs but stimulate the vasodilation mediated by the ETB1 receptor [28].
3.3. PGI2 Pathway
Prostacyclin (PGI2) is a lipidic mediator in endothelial cells. PGI2 is produced by prostacyclin sinthase through the degradation of arachidonic acid in endothelial cells (Figure 1). PGI2 is a potent pulmonary vasodilator mediator released by endothelial cells through the concerted actions of the cyclooxygenase and prostacyclin synthase. Using arachidonic acid and prostaglandin H2 as a substrate, PGI2 promotes pulmonary vasodilation and has antithrombotic and antiproliferative properties (Figure 1) [29]. The effects of PGI2 occur through the I-prostanoid receptor (IPR) via the activation of adenylyl cyclase, thereby converting ATP to cAMP, which increases protein kinase A (PKA) activity. In turn, PKA promotes the phosphorylation of myosin light chain kinase, which leads to smooth muscle relaxation and vasodilation (Figure 1). PKA promotes arterial relaxation by (a) phosphorylating myosin light chain kinase at a site that reduces the affinity of the kinase to the calcium–calmodulin complex, (b) activating calcium pumps in the cell membrane and sarcoplasmic reticulum, and (c) opening potassium channels through which potassium can exit and thereby hyperpolarize the cell [30].
IPRs are expressed in the VSMCs of the pulmonary vasculature [31][32]. In PAH patients, the PGI2 levels are reduced in the pulmonary vasculature and serum. IPRs are also reduced in patients with PAH [33][34]. Prostanoid drugs are synthetic analogs of IP that can be administered in oral, inhaled, subcutaneous, or intravenous forms. Epoprostenol, Treprostinil, and selexipag are drugs of this group that have demonstrated positive effects in the treatment of PAH [35][36][37][38][39][40][41].
Treatments for PAH are mainly focused on blocking the pathophysiological mechanisms and the activation of vasodilatory mechanisms. However, PAH is complex and multifactorial. Therefore, the ideal treatment should be focused on blocking/stimulating more than one mechanism. Indeed, in clinical practice, cotherapy and combination therapy are becoming the basis for the successful management of PAH [42]. Recently, new drugs have been introduced into clinical practice, all targeting endothelin, nitric oxide, and prostacyclin pathways, with the main objective of improving function, quality of life, and increasing survival. However, there are still a significant number of patients that do not respond adequately to or tolerate such medical therapies. Moreover, none of the available therapies have been shown to slow the progression of the disease [27][43]. On the other hand, interest in the search for the mechanisms involved and, therefore, therapeutic options continue.
References
- Gérald Simonneau; David Montani; David S Celermajer; Christopher P. Denton; Michael A. Gatzoulis; Michael Krowka; Paul G. Williams; Rogerio Souza; Haemodynamic definitions and updated clinical classification of pulmonary hypertension. European Respiratory Journal 2019, 53, 1801913, 10.1183/13993003.01913-2018.
- Wouter Jacobs; Mariëlle C. Van De Veerdonk; Pia Trip; Frances De Man; Martijn W. Heymans; Johannes T. Marcus; Steven M. Kawut; Harm-Jan Bogaard; Anco Boonstra; Anton Vonk Noordegraaf; et al. The Right Ventricle Explains Sex Differences in Survival in Idiopathic Pulmonary Arterial Hypertension. Chest 2014, 145, 1230-1236, 10.1378/chest.13-1291.
- Nathan Hatton; John J Ryan; Sex differences in response to pulmonary arterial hypertension therapy: is what's good for the goose, good for the gander?. Chest 2014, 145, 1184-1186, 10.1378/chest.13-3061.
- Marc Humbert; Christophe Guignabert; Sébastien Bonnet; Peter Dorfmüller; James R. Klinger; Mark R. Nicolls; Andrea J. Olschewski; Soni Savai Pullamsetti; Ralph Theo Schermuly; Kurt R. Stenmark; et al.Marlene Rabinovitch Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. European Respiratory Journal 2019, 53, 1801887, 10.1183/13993003.01887-2018.
- Christophe Guignabert; Ly Tu; Barbara Girerd; Nicolas Ricard; Alice Huertas; David Montani; Marc Humbert; New Molecular Targets of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Chest 2015, 147, 529-537, 10.1378/chest.14-0862.
- Alice Huertas; Frédéric Perros; Ly Tu; Sylvia Cohen-Kaminsky; David Montani; Peter Dorfmüller; Christophe Guignabert; Marc Humbert; Immune Dysregulation and Endothelial Dysfunction in Pulmonary Arterial Hypertension: A Complex Interplay. Circulation 2014, 129, 1332-1340, 10.1161/circulationaha.113.004555.
- U S Ryan; J W Ryan; Cell biology of pulmonary endothelium.. Circulation 1984, 70, 11146.
- U S Ryan; Pulmonary endothelium: a dynamic interface.. Clinical and Investigative Medicine 1985, 9, 124-32.
- Alice Huertas; Christophe Guignabert; Joan A. Barberà; Peter Bärtsch; Jahar Bhattacharya; Sunita Bhattacharya; Maria R. Bonsignore; Laurence Dewachter; Anh Tuan Dinh-Xuan; Peter Dorfmüller; et al.Mark T. GladwinMarc HumbertTom KotsimbosTheodoros VassilakopoulosOlivier SanchezLaurent SavaleUgo TestaMartin R. Wilkins Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases. European Respiratory Journal 2018, 51, 1700745, 10.1183/13993003.00745-2017.
- Vishal Parikh; Anju Bhardwaj; Ajith Nair; Pharmacotherapy for pulmonary arterial hypertension.. Journal of Thoracic Disease 2019, 11, S1767-S1781, 10.21037/jtd.2019.09.14.
- David A Kass; Takahiro Nagayama; Eiki Takimoto; Hunter C. Champion; Phosphodiesterase regulation of nitric oxide signaling. Cardiovascular Research 2007, 75, 303-314, 10.1016/j.cardiores.2007.02.031.
- Jayan Nagendran; S L Archer; Daniel Soliman; Vikram Gurtu; Rohit Moudgil; Alois Haromy; Chantal St. Aubin; Linda Webster; Ivan M. Rebeyka; David B. Ross; et al.Peter E LightJason R.B. DyckEvangelos D. Michelakis Phosphodiesterase Type 5 Is Highly Expressed in the Hypertrophied Human Right Ventricle, and Acute Inhibition of Phosphodiesterase Type 5 Improves Contractility. Circulation 2007, 116, 238-248, 10.1161/circulationaha.106.655266.
- Jackie D. Corbin; Sharron H. Francis; Molecular biology and pharmacology of PDE-5-inhibitor therapy for erectile dysfunction.. Journal of Andrology 2003, 24, S38-S41, 10.1002/j.1939-4640.2003.tb02744.x.
- Johannes-Peter Stasch; Pal Pacher; Oleg V. Evgenov; Soluble Guanylate Cyclase as an Emerging Therapeutic Target in Cardiopulmonary Disease. Circulation 2011, 123, 2263-2273, 10.1161/CIRCULATIONAHA.110.981738.
- Johannes-Peter Stasch; Oleg V. Evgenov; Soluble Guanylate Cyclase Stimulators in Pulmonary Hypertension. Drug Delivery 2013, 218, 279-313, 10.1007/978-3-642-38664-0_12.
- Nazzareno Galiè; Katharina Müller; Andrea-Viviana Scalise; Ekkehard Grünig; PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. European Respiratory Journal 2015, 45, 1314-1322, 10.1183/09031936.00105914.
- John M Tovar; John G Gums; Tezosentan in the Treatment of Acute Heart Failure. Annals of Pharmacotherapy 2003, 37, 1877-1883, 10.1345/aph.1d080.
- Lewis J. Rubin; Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sciences 2012, 91, 517-521, 10.1016/j.lfs.2012.07.033.
- Lyn R. Frumkin; The Pharmacological Treatment of Pulmonary Arterial Hypertension. Pharmacological Reviews 2012, 64, 583-620, 10.1124/pr.111.005587.
- Y Hirata; T Emori; S Eguchi; K Kanno; T Imai; K Ohta; F Marumo; Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells.. Journal of Clinical Investigation 1993, 91, 1367-1373, 10.1172/jci116338.
- Satoru Eguchi; Yukio Hirata; Fumiaki Marumo; Endothelin Subtype B Receptors Are Coupled to Adenylate Cyclase via Inhibitory G Protein in Cultured Bovine Endothelial Cells. Journal of Cardiovascular Pharmacology 1992, 22, S161-S163, 10.1097/00005344-199322008-00043.
- Frederik C. Enevoldsen; Jayashree Sahana; Markus Wehland; Daniela Grimm; Manfred Infanger; Marcus Krüger; Endothelin Receptor Antagonists: Status Quo and Future Perspectives for Targeted Therapy. Journal of Clinical Medicine 2020, 9, 824, 10.3390/jcm9030824.
- Adel Giaid; Masashi Yanagisawa; David Langleben; Rene P. Michel; Robert Levy; Hani Shennib; Sadao Kimura; Tomoh Masaki; William P. Duguid; Duncan Stewart; et al. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. New England Journal of Medicine 1993, 328, 1732-1739, 10.1056/nejm199306173282402.
- Neil Davie; Stephen J. Haleen; Paul D. Upton; Julia M. Polak; Magdi H. Yacoub; Nicholas W Morrell; John Wharton; ETAand ETBReceptors Modulate the Proliferation of Human Pulmonary Artery Smooth Muscle Cells. American Journal of Respiratory and Critical Care Medicine 2002, 165, 398-405, 10.1164/ajrccm.165.3.2104059.
- L.J. Rubin; D.B. Badesch; R.J. Barst; Bosentan therapy for pulmonary arterial hypertension. ACC Current Journal Review 2002, 11, 30, 10.1016/s1062-1458(02)00782-1.
- Nazzareno Galiè; Lj Rubin; Mm Hoeper; Pavel Jansa; H Al-Hiti; Gmb Meyer; E Chiossi; A Kusic-Pajic; G. Simonneau; Marius M. Hoeper; et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. The Lancet 2008, 371, 2093-2100, 10.1016/s0140-6736(08)60919-8.
- Tomás Pulido; Igor Adzerikho; Richard N. Channick; Marion Delcroix; Nazzareno Galié; Hossein Ardeschir Ghofrani; Pavel Jansa; Zhi-Cheng Jing; Franck-Olivier Le Brun; Sanjay Mehta; et al.Camilla M. MittelholzerLoïc PerchenetB.K.S. SastryOlivier SitbonRogerio SouzaAdam TorbickiXiaofeng ZengLewis J. RubinGérald Simonneau Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. New England Journal of Medicine 2013, 369, 809-818, 10.1056/nejmoa1213917.
- M.-C. Chaumais; Christophe Guignabert; Laurent Savale; Xavier Jaïs; Athénaïs Boucly; David Montani; Gerald Simonneau; Marc Humbert; Olivier Sitbon; Clinical Pharmacology of Endothelin Receptor Antagonists Used in the Treatment of Pulmonary Arterial Hypertension. American Journal of Cardiovascular Drugs 2014, 15, 13-26, 10.1007/s40256-014-0095-y.
- J Vane; R.E Corin; Prostacyclin: A Vascular Mediator. European Journal of Vascular and Endovascular Surgery 2003, 26, 571-578, 10.1016/s1078-5884(03)00385-x.
- P. Macke Consigny; Vascular Smooth Muscle Contraction and Relaxation: Pathways and Chemical Modulation. Journal of Vascular and Interventional Radiology 1991, 2, 309-317, 10.1016/s1051-0443(91)72249-5.
- Rüdiger Blindt; Anja K. Bosserhoff; Jürgen Vom Dahl; Peter Hanrath; Karsten Schrör; Thomas Hohlfeld; Jutta Meyer-Kirchrath; Activation of IP and EP(3) receptors alters cAMP-dependent cell migration.. European Journal of Pharmacology 2002, 444, 31-37, 10.1016/s0014-2999(02)01607-2.
- John Wharton; Neil Davie; Paul. D. Upton; Magdi H. Yacoub; Julia M. Polak; Nicholas W Morrell; Prostacyclin analogues differentially inhibit growth of distal and proximal human pulmonary artery smooth muscle cells.. Circulation 2000, 102, 3130-3136, 10.1161/01.cir.102.25.3130.
- Brian W. Christman; Charles D. McPherson; John H. Newman; Gayle A. King; Gordon R. Bernard; Bertron M. Groves; James E. Loyd; An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary Hypertension. New England Journal of Medicine 1992, 327, 70-75, 10.1056/nejm199207093270202.
- Rubin M. Tuder; Carlyne D. Cool; Mark W. Geraci; Jun Wang; Steven H. Abman; Laurel Wright; David Badesch; Norbert F. Voelkel; Prostacyclin Synthase Expression Is Decreased in Lungs from Patients with Severe Pulmonary Hypertension. American Journal of Respiratory and Critical Care Medicine 1999, 159, 1925-1932, 10.1164/ajrccm.159.6.9804054.
- Robyn J. Barst; Lewis J. Rubin; Walker A. Long; Michael D McGoon; Stuart Rich; David Badesch; Bertron M. Groves; Victor F. Tapson; Robert C. Bourge; Bruce H. Brundage; et al.Spencer K. KoernerDavid LanglebenCesar A. KellerSrinivas MuraliBarry F. UretskyLinda M. ClaytonMaria M. JöbsisShelmer D. BlackburnDenise ShortinoJames W. Crow A Comparison of Continuous Intravenous Epoprostenol (Prostacyclin) with Conventional Therapy for Primary Pulmonary Hypertension. New England Journal of Medicine 1996, 334, 296-301, 10.1056/nejm199602013340504.
- Gérald Simonneau; Robyn J. Barst; Nazzareno Galiè; Robert Naeije; Stuart Rich; Robert C. Bourge; Anne Keogh; Ronald Oudiz; Adaani Frost; Shelmer D. Blackburn; et al.James W. CrowLewis J. Rubin Continuous Subcutaneous Infusion of Treprostinil, a Prostacyclin Analogue, in Patients with Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine 2002, 165, 800-804, 10.1164/ajrccm.165.6.2106079.
- Vallerie V. McLaughlin; Raymond Benza; Lewis J. Rubin; Richard N. Channick; R. Voswinckel; Victor F. Tapson; Ivan M. Robbins; Horst Olschewski; Melvyn Rubenfire; Werner Seeger; et al. Addition of Inhaled Treprostinil to Oral Therapy for Pulmonary Arterial Hypertension. Journal of the American College of Cardiology 2010, 55, 1915-1922, 10.1016/j.jacc.2010.01.027.
- H. Olschewski; G. Simonneau; N. Galiè; Inhaled iloprost for severe pulmonary hypertension. ACC Current Journal Review 2002, 11, 19, 10.1016/s1062-1458(02)00905-4.
- Victor F. Tapson; Zhi-Cheng Jing; Kai-Feng Xu; Lei Pan; Jeremy Feldman; David G. Kiely; Eugene Kotlyar; C. Shane McSwain; Kevin Laliberte; Carl Arneson; et al.Lewis J. Rubin Oral Treprostinil for the Treatment of Pulmonary Arterial Hypertension in Patients Receiving Background Endothelin Receptor Antagonist and Phosphodiesterase Type 5 Inhibitor Therapy (The FREEDOM-C2 Study). Chest 2013, 144, 952-958, 10.1378/chest.12-2875.
- V.F. Tapson; C.J. Sanchez Diaz; G.M. Bohns Meyer; T. Pulido; P. Sepulveda; K.Y. Wang; C.Q. Deng; R. Grover; D. Solum; A. Ousmanou; et al.R.J. White Treatment with Oral Treprostinil Delays Time to Clinical Worsening in Patients with Pulmonary Arterial Hypertension - Results from FREEDOM-EV. The Journal of Heart and Lung Transplantation 2019, 38, S94-S95, 10.1016/j.healun.2019.01.220.
- G. Simonneau; Karol Adam Kamiński; Marius M. Hoeper; Marion Delcroix; Kristóf Karlócai; Nazzareno Galiè; Bruno Degano; Diana Bonderman; Marcin Kurzyna; Michela Efficace; et al.Ruben GiorginoIrene M. Lang Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. European Respiratory Journal 2012, 40, 874-880, 10.1183/09031936.00137511.
- James R. Klinger; C. Gregory Elliott; Deborah J. Levine; Eduardo Bossone; Laura Duvall; Karen Fagan; Julie Frantsve-Hawley; Steven M. Kawut; John J. Ryan; Erika B. Rosenzweig; et al.Nneka SederstromVirginia D. SteenDavid BadeschGregory Elliott Therapy for Pulmonary Arterial Hypertension in Adults. Chest 2019, 155, 565-586, 10.1016/j.chest.2018.11.030.
- David L. Prior; Heath Adams; Trevor J Williams; Update on pharmacotherapy for pulmonary hypertension. Medical Journal of Australia 2016, 205, 271-276, 10.5694/mja16.00468.


