Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ralf Weiskirchen | + 3018 word(s) | 3018 | 2022-01-14 04:59:52 | | | |
| 2 | Camila Xu | -957 word(s) | 2061 | 2022-01-17 02:38:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Weiskirchen, R. Autophagy in Parenchymal and Non-Parenchymal Liver Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/18259 (accessed on 03 August 2026).
Weiskirchen R. Autophagy in Parenchymal and Non-Parenchymal Liver Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/18259. Accessed August 03, 2026.
Weiskirchen, Ralf. "Autophagy in Parenchymal and Non-Parenchymal Liver Cells" Encyclopedia, https://encyclopedia.pub/entry/18259 (accessed August 03, 2026).
Weiskirchen, R. (2022, January 14). Autophagy in Parenchymal and Non-Parenchymal Liver Cells. In Encyclopedia. https://encyclopedia.pub/entry/18259
Weiskirchen, Ralf. "Autophagy in Parenchymal and Non-Parenchymal Liver Cells." Encyclopedia. Web. 14 January, 2022.
Copy Citation
Autophagy is a highly conserved intracellular process for the ordered degradation and recycling of cellular components in lysosomes. In the liver this process is relevant for maintaining liver homeostasis, especially in conditions of hepatic insults.
hepatocytes
hepatic stellate cells
sinusoidal endothelial cells
macrophages
1. Introduction
The term autophagy summarizes the processes involved in the orderly degradation and recycling of worn, abnormal, or malfunctional cellular components. It is commonly accepted today that the term “autophagy” was first introduced in 1963 by the Belgian cytologist and biochemistry Christian René de Duve, who also coined the terms “endocytosis” and “phagocytosis” to designate pathways bringing substrates for digestion in lysosomes [1]. However, the terms autophagy/autophagy/autophagia were in fact already used a century earlier and published in 1859 in a French journal [2]. The importance of autophagy was prominently acknowledged in 2016, when Yoshinori Ohsumi was awarded the Nobel Prize for Physiology or Medicine for his discoveries of mechanisms for autophagy. Autophagy is nowadays considered as a dynamic recycling system, which is essential for cellular renovation and homeostasis [3]. As such, the resultant degradation products can be used for new protein synthesis, energy production, and gluconeogenesis. There are three classes of autophagy, namely macroautophagy, microautophagy, and chaperone-mediated autophagy, requiring different sets of autophagy-related genes and cellular compounds [3] (Figure 1). Macroautophagy is the most prevalent form of autophagy. It is dependent on the “autophagosome”, a spherical vesicle appearing randomly throughout the cytoplasm with the capacity to traffic along microtubules towards the microtubule-organizing center, where lysosomes are concentrated [4]. These ring-shaped structures are majorly formed by the “AuTophaGy” (ATGs) genes that are evolutionarily conserved from yeast to higher eukaryotes. This cellular compartment has the capacity to sequester small portions of cytoplasm enriched in soluble materials and organelles and to fuse with lysosomes forming the autolysosome, in which the material is finally degraded. On the contrary, microautophagy is a more diverse type of autophagy, in which cytoplasmic compounds or spontaneous formed vesicles are directly engulfed by lysosomes. Recent studies demonstrate that this pathway is of particular relevance for cells under amino acid starvation [5]. Based on the finding that vascular membranes and endosomes can also incorporate or capture peroxisomes or lysosome-derived organelles, it was proposed that this autophagy branch should be classified in three distinct subtypes of microautophagy [6]. Chaperone-mediated autophagy is more selective and not associated with membrane reorganization [3]. Instead, chaperone and co-chaperone proteins recognize cytosolic proteins that carry specific peptide recognition sites and are then targeted to receptors on lysosomes, which subsequently internalize these proteins for degradation (Figure 1). This pathway majorly contributes to the maintenance of cellular homeostasis by facilitating degrading of proteins and recycling of amino acids. However, transgenic mouse models have shown that this pathway participates in the regulation of glucose and lipid metabolism, DNA repair, cellular reprogramming, and cellular response to stress [7].
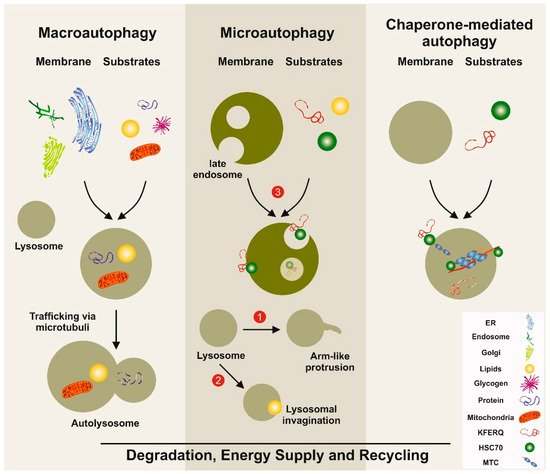
Figure 1. Simplified models of autophagy pathways in the liver. Macroautophagy involves the formation of a double-membrane vesicle, in which the substrates to be degraded are included. This vesicle called the autophagosome is then fused with the lysosome, allowing the degradation of the products. Three distinct types of microautophagy exist. In one type, the lysosome forms arm-like protrusions capable of engulfing substances. In a second branch, the lysosome can form invaginations, in which substrates (e.g., lipids) can be wrapped. The most important pathway in microautophagy involves the late endosome. In this compartment, substrates such as proteins carrying the pentapeptide lysine-phenylalanine-glutamic acid-arginine-glutamine (KFERQ)-like motifs are internalized and degraded. In chaperone-mediated autophagy, substrates with a KFERQ-like motif are first recognized by the cytosolic chaperone. Subsequently, this complex is recognized by chaperone-mediated autophagy associated receptors located at the lysosomal compartment. After internalization, the incorporated substances are degraded. The three autophagy pathways serve as a dynamic recycling system that produces new building blocks and provides energy necessary to guarantee cellular homeostasis. ER: endoplasmic reticulum; HSC70: heat-shock 70-Kd protein; MTC: multimeric translocation complex.
With regards to the liver, there is strong evidence that the process of macroautophagy in particular is the most important for maintaining hepatic homeostasis and suppressing spontaneous tumorigenesis. The systemic mosaic deletion of Atg5 in mice resulted in multiple benign tumors that developed only in the liver but not in other tissues [8]. On the other side, host-specific deletion of Atg7 impaired the growth of multiple allografted tumors in mice, most likely by inducing release of arginosuccinate synthase 1 from the liver and degradation of circulating arginine, which is essential for tumor growth [9]. These inverse findings demonstrate that autophagy plays a dual role in cancer cells with potential to both inhibit and promote tumor progression and promotion.
2. Principal Functions and Molecular Mechanisms of Autophagy
Autophagy is an important conserved recycling process necessary to maintain energy balance in the cells. In the liver, the activity of this cellular autophagy activity is enhanced or reduced in response to environmental changes and cellular needs [10]. It is not only essential for replenishing the free pool of amino acids through protein breakdown, but it also contributes to mobilization and hydrolysis of lipid stores and glycogen, thereby significantly contributing to the cellular energetics and energetic flux through different metabolic pathways [10]. The occurrence of three different types of autophagy provides a high functional variety of possible breakdown and recycling processes, which are particularly relevant for the liver, which represents the central organ in the control of organismal energy balance (Figure 1). Consequently, alteration in proper autophagy function can result in severe metabolic disorders such as obesity, fatty liver, diabetes, and other metabolic age-related disorders [11][12]. Recent findings further suggest autophagy as a critical mechanism in regulating the “liver clock” and circadian glucose metabolism by timely degrading core circadian repressor clock proteins such as crytochrome 1 (CRY1), resulting in gluconeogenesis and increased blood glucose levels [13]. Interestingly, high-fat feeding decreased CRY1 protein expression in an autophagy-dependent manner, while restoring hepatic CRY1 reversed obesity-associated hyperglycemia, suggesting that this regulatory network is a potential attractive target for therapy of obesity-associated hyperglycemia [13].
There is also first evidence that autophagy in liver aggravates the oxidative stress response during acute liver injury. In particular, autophagy maintains liver endothelial cell homeostasis and protects against cellular dysfunction, intrahepatic nitric oxide accumulation, and a liver microenvironment that promotes fibrosis [14]. Similarly, the blockade of autophagy by the autophagy inhibitor LY294002 or small interfering RNAs (siRNAs) targeting Atg5 attenuated drug-induced anti-inflammatory effects in hepatic stellate cells and on liver fibrosis [15].
Mechanistically, there is experimental evidence showing the PI3K/Akt/mTOR pathway to be critically involved in the activation of autophagy, thereby preventing cell death, promoting anticancer effects of therapeutic drugs, and reducing tumor growth [16]. On the contrary, in hepatocellular carcinoma (HCC) cells, the induction of the PI3K/Akt/mTOR pathway by α-fetoprotein (AFP) resulted in reduced cell autophagy and more malignant behavior [17]. These opposite findings demonstrate that the same autophagy-associated pathway are highly dynamic and can have pro-tumor or anti-tumor effects. Hence, the role of autophagy in HCC development is dependent on the context of liver cells, the hepatic microenvironment, stage of tumor development, or many other unrecognized factors. It is most likely that autophagy plays an anti-tumor role in normal liver cells by maintaining cell homeostasis, while it promotes the survival of HCC cells within the tumor microenvironment once the tumor is formed [18].
3. Autophagy in Homeostasis of the Liver—Implications for Hereditary Liver Diseases
The importance of autophagy for the maintenance of liver homeostasis is best exemplified in conditions, in which large quantities of misfolded proteins are formed that lead to an overburden of the proteolytic pathway involved in autophagy. Prototypically, patients suffering from classical α1-antitrypsin (α1AT) deficiency synthesize large quantities of mutant α1AT Z (ATZ) protein in which a point mutation results in a substitution of lysine for glutamate at residue 342 [19]. While the normal α1AT protein (M protein) is rapidly secreted into the blood, the missense mutation results in a polymerized mutant α1AT protein (Z protein) that is retained in the endoplasmic reticulum of hepatocytes rather than secreted in the body fluids where its physiological function is to inhibit neutrophil proteases [19][20]. Hepatocytes deal with the burden of insoluble aggregates by activating endoplasmic reticulum-associated proteasomal degradation pathways and by macroautophagy [21]. However, in most homozygous individuals these countermeasures are insufficient to overcome the overload with insoluble proteins, provoking cell death and chronic liver damage. The clinical manifestation of liver disease associated with α1AT deficiency is highly variable, and there is currently no specific treatment of α1AT-related liver disease [22]. Enhancing cellular degradation pathways, particularly autophagy, for mutant ATZ proteins may therefore represent a realistic option in the near future [23]. Independent experimental studies have shown that the induction of autophagic degradation of mutant polymerized Z protein by hepatic gene transfer of master autophagy regulators or by autophagy-enhancing drugs such as carbamazepine, rapamycin, or 24-norursodeoxycholic acid (norUDCA) can significantly reduce liver injury [21][24][25][26]. These approaches, along other targets (e.g., blocking mutant ATZ production by siRNA), are currently under clinical evaluation,
Another inherited disorder reflecting the importance of autophagy in liver homeostasis is Wilson’s disease, also known as hepatolenticular degeneration or “copper storage disease”. It represents a rare autosomal recessive disorder caused by mutation in the ATPase copper transporting protein ATP7B, preventing the body from removing excess copper and leading to accumulation of this trace metal in liver and brain [27]. Recently, it was shown that ATP7B-deficient cells showed significant increased expression of autophagy-associated genes when compared to control cells. Furthermore, hepatocytes derived from patients suffering from Wilson’s disease, as well as hepatocytes derived from Atp7b null mice and rats, contained elevated quantities of autophagosomes [28]. Interestingly, the pharmacological inhibition of ATG7 and ATG13 accelerated cell death in the hepatoma cell line HepG2 when depleted for ATP7B expression, suggesting that autophagy protects against metal toxicity and copper-induced cell death in the setting of Wilson’s disease [28].
Alcohol abuse is a third condition in which the importance of autophagy for liver homeostasis is well documented. Alcoholic liver disease (ALD) is a global healthcare problem associated with fatty liver, alcoholic hepatitis, fibrosis, and cirrhosis. During chronic ethanol consumption, the rates of autophagy are retarded in the liver, because ethanol is thought to cause faulty lysosome biogenesis and slower breakdown of lipid droplets [29]. A recent experimental study found that liver tissue from mice fed with ethanol displayed lower expression levels of total and nuclear transcription factor EB (TFEB) compared with control mice, alongside decreased parenchymal lysosome biogenesis and autophagy [30]. When the hepatic expression of the transcription factor TFEB was increased by administration of torin-1, representing an effective inducer of autophagy, or by administration of an adenoviral vector expressing TFEB, mice showed decreased steatosis and liver injury induced by ethanol, while the knock down of TFEB using an adenovirus small hairpin RNA (shRNA) approach resulted in more severe liver disease [30]. These experiments demonstrate the fundamental protective role of autophagy in formation of ALD.
Collectively, these findings from hereditary and toxic liver diseases corroborate that autophagy as a cellular degradation and clearance pathway is critical for maintaining liver homeostasis, especially in conditions of hepatic insults.
4. Autophagy in Liver Metabolism and Fatty Liver Disease
The most common liver disease worldwide is non-alcoholic fatty liver disease (NAFLD), that is characterized by extrahepatic features of the metabolic syndrome (obesity, type 2 diabetes, dyslipidemia) and distinct hepatic histological features [31]. A fraction of these patients develop non-alcoholic steatohepatitis (NASH), characterized by steatosis, inflammation, and hepatocyte ballooning, and are at a particular risk for progressing towards fibrosis, cirrhosis, and HCC [32]. Autophagy is a central “recycling mechanism” in hepatocytes, evolutionarily evolved to provide energy and to salvage key metabolites for sustaining anabolism [33]. Autophagy is therefore a key mediator of liver metabolism and is dysregulated in NAFLD [10]. For instance, autophagy provides amino acids to cellular processes via protein degradation and recycling of cell organelles [33][34], mobilizes intracellular glycogen storages (“glycophagy”) in case of starvation [33], and breaks down lipid droplets (“lipophagy”), which increases intracellular triglyceride and free fatty acid concentrations [35]. High levels of energy substrates (e.g., ATP), insulin, or free fatty acids negatively regulate autophagy, while starvation is one of the strongest physiological activators of autophagy in hepatocytes [10]. Importantly, hepatic autophagy is decreased overall in association with conditions that predispose to NAFLD such as obesity and aging [36]. Although an extensive body of literature suggests that the pharmacological modulation of either autophagy directly or autophagy-related up- or downstream pathways could hold therapeutic potential in obesity, metabolic syndrome, or NAFLD/NASH [37], lifestyle interventions including fasting, dietary changes, and exercise may also be very potent inducers of beneficial autophagy-related changes in metabolism [38][39].
References
- Sabatini, D.D.; Adesnik, M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc. Natl. Acad. Sci. USA 2013, 110, 13234–13235.
- Ktistakis, N.T. In praise of M. Anselmier who first used the term “autophagie” in 1859. Autophagy 2017, 13, 2015–2017.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741.
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, 29–34.
- Olsvik, H.L.; Svenning, S.; Abudu, Y.P.; Brech, A.; Stenmark, H.; Johansen, T.; Mejlvang, J. Endosomal microautophagy is an integrated part of the autophagic response to amino acid starvation. Autophagy 2018, 15, 182–183.
- Oku, M.; Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays 2018, 40, e1800008.
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800.
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573.
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of liver metabolism by autophagy. Gastroenterology 2016, 150, 328–339.
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200.
- Moulis, M.; Vindis, C. Autophagy in metabolic age-related human diseases. Cells 2018, 7, 149.
- Toledo, M.; Batista-Gonzalez, A.; Merheb, E.; Aoun, M.L.; Tarabra, E.; Feng, D.; Sarparanta, J.; Merlo, P.; Botrè, F.; Schwartz, G.J.; et al. Autophagy regulates the liver clock and glucose metabolism by degrading CRY1. Cell Metab. 2018, 28, 268–281.
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2018.
- Liu, Z.; Zhu, P.; Zhang, L.; Xiong, B.; Tao, J.; Guan, W.; Li, C.; Chen, C.; Gu, J.; Duanmu, J.; et al. Autophagy inhibition attenuates the induction of anti-inflammatory effect of catalpol in liver fibrosis. Biomed. Pharmacother. 2018, 103, 1262–1271.
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707.
- Wang, S.; Zhu, M.; Wang, Q.; Hou, Y.; Li, L.; Weng, H.; Zhao, Y.; Chen, D.; Ding, H.; Guo, J.; et al. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2018, 9, 1027.
- Liu, L.; Liao, J.Z.; He, X.X.; Li, P.Y. The role of autophagy in hepatocellular carcinoma: Friend or foe. Oncotarget 2017, 8, 57707–57722.
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, 851–862.
- Teckman, J.H.; Blomenkamp, K.S. Pathophysiology of alpha-1 antitrypsin deficiency liver disease. Methods Mol. Biol. 2017, 1639, 1–8.
- Tang, Y.; Blomenkamp, K.S.; Fickert, P.; Trauner, M.; Teckman, J.H. NorUDCA promotes degradation of α1-antitrypsin mutant Z protein by inducing autophagy through AMPK/ULK1 pathway. PLoS ONE 2018, 13, e0200897.
- Patel, D.; Teckman, J.H. Alpha-1-antitrypsin deficiency liver disease. Clin. Liver Dis. 2018, 22, 643–655.
- Tacke, F.; Trautwein, C. Controlling autophagy: A new concept for clearing liver disease. Hepatology 2011, 53, 356–358.
- Pastore, N.; Blomenkamp, K.; Annunziata, F.; Piccolo, P.; Mithbaokar, P.; Maria Sepe, R.; Vetrini, F.; Palmer, D.; Ng, P.; Polishchuk, E.; et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 2013, 5, 397–412.
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232.
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. 2010, 235, 700–709.
- Weiskirchen, S.; Kim, P.; Weiskirchen, R. Determination of copper poisoning in Wilson’s disease using laser ablation inductively coupled plasma spectrometry. Ann. Transl. Med. 2018.
- Polishchuk, E.V.; Merolla, A.; Lichtmannegger, J.; Romano, A.; Indrieri, A.; Ilyechova, E.Y.; Concilli, M.; De Cegli, R.; Crispino, R.; Mariniello, M.; et al. Activation of autophagy, observed in liver tissues from patients with Wilson disease and from Atp7b-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology 2018.
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic liver disease: Pathogenesis and current management. Alcohol Res. 2017, 38, 147–161.
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology 2018, 155, 865–879.
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global perspectives on non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Hepatology 2018.
- Diehl, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072.
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472.
- Lamming, D.W.; Bar-Peled, L. Lysosome: The metabolic signaling hub. Traffic 2019, 20, 27–38.
- Schulze, R.J.; Drižytė, K.; Casey, C.A.; McNiven, M.A. Hepatic lipophagy: New insights into autophagic catabolism of lipid droplets in the liver. Hepatol. Commun. 2017, 1, 359–369.
- Czaja, M.J. Function of autophagy in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2016, 61, 1304–1313.
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376.
- Van Niekerk, G.; du Toit, A.; Loos, B.; Engelbrecht, A.M. Nutrient excess and autophagic deficiency: Explaining metabolic diseases in obesity. Metabolism 2018, 82, 14–21.
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–555.
More
Information
Subjects:
Gastroenterology & Hepatology; Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
17 Jan 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No