 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Svenja Koslowski | + 2214 word(s) | 2214 | 2020-07-02 11:19:40 | | | |
| 2 | Nora Tang | Meta information modification | 2214 | 2020-07-06 08:54:40 | | | | |
| 3 | Nora Tang | Meta information modification | 2214 | 2020-07-06 08:55:28 | | |
Video Upload Options
Autosomal dominant polycystic kidney disease (ADPKD) is a complex monogenic kidney disease which progressively leads to kidney failure. A brief description of the main characteristics and known causes of ADPKD pathology, as well as the current pharmacological treatment is presented. Numerous research tools have been developed over the last decades to study this pathology and have allowed significant progress in understanding ADPKD specific disease mechanisms, even though the precise pathophysiological mechanisms of this pathology still remain elusive. As no perfect model of ADPKD currently exists, a broad overview of available and emerging in vitro and in vivo research models used to study this disease is given. Finally, a multimodal approach, combining different and carefully selected in vitro and in vivo research tools is suggested, in order to produce reliable results relevant to the human disease.
1. Definition
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic inherited kidney disease in humans and the leading inheritable cause of end stage renal disease (ESRD). Its main symptoms are the progressive formation and enlargement of fluid filled renal cysts which gradually impair kidney function until renal replacement therapy, such as dialysis or kidney transplantation becomes necessary [1][2][3][4][5]. ADPKD patients present highly enlarged kidneys, which can reach a total volume exceeding 3500 mL for both kidneys combined [6] (versus a mean single kidney volume of 196 mL in healthy individuals [7]). Important kidney enlargement can sometimes be observed even before a significant decline in kidney function. Notably, ADPKD patients account for about 6% of all patients under renal replacement therapy [8]. In addition, numerous extra-renal manifestations of the disease, such as pancreatic and liver cysts, increased blood pressure, and a higher risk of cerebral aneurisms have been reported [9].
2. Introduction
As of today, the selective vasopressin V2 receptor antagonist tolvaptan (Jinarc® (EU, UK, Canada), Jynarque® (USA), Samsca® (Japan)) is the only available treatment that can slow cyst growth in ADPKD patients [10][11]. Through the inhibition of the binding of the antidiuretic hormone arginine vasopressin (AVP) to its receptor, tolvaptan diminishes cAMP production and thus cAMP-associated cyst cell proliferation and cyst fluid secretion. However, this treatment is only approved in adult patients at risk or showing evidence of rapidly progressing disease. Furthermore, it is not suitable for all patients due to its risk of hepatotoxicity and some other adverse effects, like polyuria and increased thirst, that can significantly lower the quality of life of people under treatment [12][13].
At the molecular level, ADPKD is mostly caused by mutations in either of two genes, PKD1 or PKD2, which encode respectively polycystin-1 (PC-1) and polycystin-2 (PC-2) [14][15][16]. A third mutated (monoallelic) gene, DNAJB11, was recently identified to trigger ADPKD [17]. Mutations in PKD1 are present in about 85% of ADPKD patients and are related to a more severe disease progression with earlier onset of end stage renal disease [1]. No human case of homozygous PKD gene mutations is known and studies in animal models suggest that the PKD genes play a crucial role during embryogenesis as homozygous knock-out mice present severe developmental defects and die before birth. Even though the precise pathophysiological mechanisms of ADPKD still remain elusive, extensive research has offered valuable insights regarding the function of the polycystin proteins (Figure 1, for review see [18]). Hence, PC-1 and PC-2 have been shown to interact at the primary cilium of renal epithelia cells, where they form a mechanosensitive cation channel, and ciliary defects appear to be implicated in ADPKD pathology. However, the polycystin proteins are also found in other subcellular locations and seem to be implicated in numerous cell signaling pathways regulating gene expression, cellular differentiation and proliferation, as well as apoptosis [19][20].
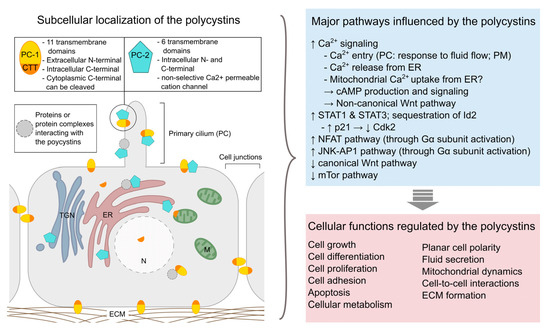
Figure 1. Overview of the subcellular localization of polycystins (left) and examples of major pathways and cellular functions affected by these proteins (right). PC-1: polycystin-1; PC-2: polycystin-2; CTT: C-terminal tail (of polycystin-1); ECM: extracellular matrix; ER: endoplasmic reticulum; M: mitochondrion; PC: primary cilium; PM: plasma membrane; TNG: trans-Golgi network. [19][21][22][23][24][25][26].
The level of functional polycystin proteins produced in the cell seems to be a crucial factor for renal cyst formation and decreased expression of the PKD genes can lead to ADPKD development in humans as well as animal models of the disease [27][28]. Importantly, renal injury seems to accelerate disease progression through the activation of repair mechanisms [29][30][31].
3. ADPKD research models
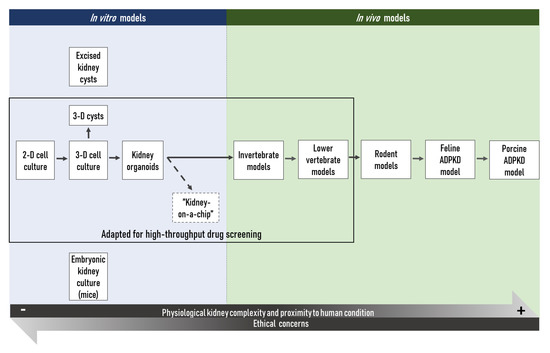
Figure 2. Schematic overview on the different ADPKD research models available to the scientific community. 2D: two-dimensional; 3D: three-dimensional.
This large choice of models can, however, be confusing and, since no perfect model of ADPKD exists to date, the suitability of a chosen model in view of a specific research hypothesis must be thoroughly considered. This issue is currently partly addressed by the multimodal approach most research teams choose. Importantly, the choice of such an approach is the result of a balance between a rigorous scientific strategy and the available resources, with the final aim to validate results in several distinct experimental conditions, ethical concerns, as well as economic considerations, not to mention the need to obtain exploitable results in a reasonable timeframe.
Most in vitro techniques present the advantages of being relatively rapid, cost-effective, and independent from any ethical issues (as compared to in vivo models). Hence, a large body of reproducible results can be rapidly obtained through standardized protocols and manipulation techniques. The main in vitro tools used in ADPKD research today are 2-dimensional or 3-dimensional cell cultures conducted with primary or immortalized kidney cell lines [32][33][34][35][36][37]. Furthermore, kidney organoids established from (patient-derived or genetically modified) pluripotent stem cells (PSCs) allow researchers to study disease mechanisms and the efficiency of potential therapeutic compounds in a more organ-like context [38][39][40][41][42]. However, even though the ‘kidney-on-a-chip’ might represent an attractive solution to a certain extent [43], in vitro models in use today are studied by essence in a complete absence of any physiological context. This may be viewed as an advantage for the investigation of isolated cellular mechanisms, but those models cannot give any indications on complex in vivo tissue or organ interactions, compound metabolization or potential off-target in vivo toxicity of a parent compound or of its metabolites. Small and widely-used invertebrate models, like the fruit fly Drosophila melanogaster and the worm Caenorhabditis elegans, and non-mammalian vertebrate models, like the zebrafish Danio rerio or the frogs of the Xenopus genus, may be considered as they represent an interesting alternative and compromise between in vitro models and complex in vivo models [44][45]. Indeed, their housing costs are relatively low, their numerous, and by definition ex utero developing embryos are, at least up to certain developmental states in the case of vertebrates, not concerned to the legislation on the protection of animals used for scientific purposes [46]. In addition, as the embryos and larvae of those animals are often transparent, they allow direct observation of physiological processes and facilitate the development of high-throughput technologies to test the toxicity or efficacy of a large number of compounds. Furthermore, an increasing number of tools for the genetic manipulation of such organisms are available nowadays. The genomes of these small model organisms have been sequenced, which allows the study of protein expression and function in a physiological context and thereby to create human disease models in these model organisms. Importantly, in biomedical research, high-throughput screening using in vitro or simple in vivo models is often the first step to identify potential therapeutic molecules. For ADPKD research, high-throughput platforms using 3D cysts [37] or kidney organoids derived from PKD knock-out human pluripotent stem cells [47], as well as the automated imaging pipeline using zebrafish larvae [48] in combination with Pkd1a/b or Pkd2 morpholino oligos could be particularly useful to detect promising candidates, while limiting false positives. The efficiency and lack of toxicity of the potential therapeutic molecules identified in such a way must then be validated in animal models more closely related to humans, such as the commonly used rodent models. Those species have again the advantage that housing costs are relatively low compared to larger mammals such as dogs or mini-pigs, while producing a relatively high number of offspring [49]. Furthermore, those organisms are very well studied and the scientific community has a large number of different transgenic or non-transgenic strains readily available. However, even though rodents are more closely related to humans than the aforementioned organisms and despite the fact that a considerable number of transgenic mouse models of human diseases exists today, results obtained in those models are not always fully transposable to humans. In addition, although murine models have proven reliable models of aging, in ADPKD research notably, the short lifespan of those animals might be a limiting factor. The vast majority of human ADPKD patients present a heterozygous mutation in one of the PKD genes and the disease progresses over decades before renal function is impaired to a point that renal replacement therapy is necessary; while in murine models, heterozygous Pkd mutations—which better recapitulate the human disease—cause a mild and very variable disease phenotype [50]. Somatic mutations of the second non-mutated PKD allele are supposed to play a role in the initiation of cyst formation in ADPKD patients [51], and even though somatic mutation rates may differ between organisms, the short lifespan of rodents might thus explain the only very mild phenotype observed as a consequence of heterozygous Pkd mutations [18][52]. In this sense, the ADPKD cat [53][54][55][56][57] and transgenic mini-pig [58][59] models seem to recapitulate human ADPKD pathogenesis more accurately than rodent models. Porcine models of human disease seem especially promising in view of their strong physiological similarity to humans [60][61]. However, up to now, only few research facilities have the required equipment, staff experienced in handling, and an adequate husbandry to house those species, not to forget that the housing costs of such models are much higher. However, in the long term, increasing use of specific mini-pig models of human disease in biomedical research might eventually decrease these expenses. In addition, the significant financial loss due to the clinical trials of molecules which seemed effective and promising in preclinical rodent models, but that later turned to be ineffective or to present important adverse effects in humans may also be in favor of the use of cat and mini-pig models for ADPKD. Examples of such molecules that showed promising results in preclinical studies carried out on rodent models of polycystic kidney disease (PKD) are the mTOR inhibitors sirolimus (rapamycin) and everolimus. While both molecules efficiently slowed down disease progression in the Han:SPRD cy/+ rat [62][63][64] and rapamycin further proved to be efficient in the bpk mouse and the orpk-rescue mouse [65], no beneficial effect of rapamycin or everolimus treatment could be observed in clinical trials with ADPKD patients [66][67]. In this context, it may be interesting to note that none of the PKD rodent models employed during the preclinical evaluation of those compounds was based on mutations in the Pkd genes.
In line, Hester Happé and Dorien J. M. Peters suggested [28] that ADPKD candidate treatments should be tested in at least two different animal models, of which at least one should be based on a PKD gene mutation. Vasopressin V2 receptor antagonists OPC-31260 and its derivative tolvaptan (OPC-41061) have for example shown effectiveness in several distinct rodent models of PKD, among those the PCK rat, the pcy mouse, the Pkd2 WS25 mouse, and a conditional kidney specific Pkd1 knock-out model [68][69][70][71], as well as in the cpk mouse [72], a model of autosomal recessive PKD (ARPKD), before and while clinical testing for its effectiveness in ADPKD treatment was conducted. However, before tests are carried out in animals, potential therapeutic compounds have to be identified in adequate in vitro models in order to limit the wasteful use of laboratory animals.
The existence of such numerous tools and models to conduct ADPKD research results from the very active research that has been carried throughout the last decades. The valuable findings which have been collected through the use of those tools help to better understand this complex disease and have made the discovery of the first drug treatment permitting to slow down ADPKD progression possible. Furthermore, the existence of several in vitro and in vivo models for this human disorder provides researchers with the flexibility to choose among the diverse models those which best suit their specific research questions. However, as of today no model perfectly recapitulates the human disease, special attention has to be paid to the specific characteristics of each model when establishing a research protocol. Finally, the InterMOD project (MOD: Model Organism Database), which “aims to make it easier to carry out cross-MOD comparative analysis and to relate MOD data to medical research” [73] should also be mentioned here. Different MODs, among those FlyMine (https://www.flymine.org/flymine/begin.do), ZebrafishMine (http://www.zebrafishmine.org/begin.do), MouseMine (http://www.mousemine.org/mousemine/begin.do), RatMine (http://ratmine.mcw.edu/ratmine/begin.do) and HumanMine (https://www.humanmine.org/humanmine/begin.do) participate to this project and thus permit cross-species analysis of biological data. Therefore, this project constitutes a helpful and interesting tool for the integration and translation of model organism research into medical applications, as well as for the comparison of model organisms in view of a specific research project.
References
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301.
- Grantham, J.J. Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2008, 359, 1477–1485.
- Brunelli, S.; Blanchette, C.; Claxton, A.; Roy, D.; Rossetti, S.; Gutierrez, B. End-stage renal disease in autosomal dominant polycystic kidney disease: A comparison of dialysis-related utilization and costs with other chronic kidney diseases. Clin. Outcomes Res. 2015, 7, 65.
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50.
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23.
- Grantham, J.J.; Torres, V.E.; Chapman, A.B.; Guay-Woodford, L.M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Baumgarten, D.A.; Kenney, P.J.; Harris, P.C.; et al. Volume Progression in Polycystic Kidney Disease. N. Engl. J. Med. 2006, 354, 2122–2130.
- van den Dool, S.W.; Wasser, M.N.; de Fijter, J.W.; Hoekstra, J.; van der Geest, R.J. Functional Renal Volume: Quantitative Analysis at Gadolinium-enhanced MR Angiography—Feasibility Study in Healthy Potential Kidney Donors. Radiology 2005, 236, 189–195.
- Spithoven, E.M.; Kramer, A.; Meijer, E.; Orskov, B.; Wanner, C.; Caskey, F.; Collart, F.; Finne, P.; Fogarty, D.G.; Groothoff, J.W.; et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014, 86, 1244–1252.
- Luciano, R.L.; Dahl, N.K. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): Considerations for routine screening and management. Nephrol. Dial. Transpl. 2014, 29, 247–254.
- Xie, X.; Cai, Q.; Guo, X.-Y.; Bai, D.-H.; Sheng, H.-Z.; Wang, B.-K.; Yan, K.; Lu, A.-M.; Wang, X.-R. Effectiveness of Tolvaptan in the Treatment for Patients with Autosomal Dominant Polycystic Kidney Disease: A Meta-analysis. Comb. Chem. High Throughput Screen 2020, 23, 6–16.
- Blair, H.A. Tolvaptan: A Review in Autosomal Dominant Polycystic Kidney Disease. Drugs 2019, 79, 303–313.
- Sans-Atxer, L.; Joly, D. Tolvaptan in the treatment of autosomal dominant polycystic kidney disease: Patient selection and special considerations. Int. J. Nephrol. Renov. Dis. 2018, 11, 41–51.
- Cowley, B.D., Jr. Polycystic Kidney Disease Progression: Learning from Europe. Am. J. Nephrol. 2018, 48, 306–307.
- Hughes, J.; Ward, C.J.; Peral, B.; Aspinwall, R.; Clark, K.; San Millán, J.L.; Gamble, V.; Harris, P.C. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 1995, 10, 151–160.
- The International Polycystic Kidney Disease Consortium. Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell 1995, 81, 289–298.
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342.
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.-P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844.
- Ong, A.C.M.; Harris, P.C. A polycystin-centric view of cyst formation and disease: The polycystins revisited. Kidney Int. 2015, 88, 699–710.
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710.
- Saigusa, T.; Bell, P.D. Molecular Pathways and Therapies in Autosomal-Dominant Polycystic Kidney Disease. Physiology 2015, 30, 195–207.
- Torres, V.E.; Harris, P.C. Mechanisms of Disease: Autosomal dominant and recessive polycystic kidney diseases. Nat. Clin. Pr. Nephrol. 2006, 2, 40–55.
- Mangos, S.; Lam, P.; Zhao, A.; Liu, Y.; Mudumana, S.; Vasilyev, A.; Liu, A.; Drummond, I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model. Mech. 2010, 3, 354–365.
- Mekahli, D.; Parys, J.B.; Bultynck, G.; Missiaen, L.; De Smedt, H. Polycystins and cellular Ca2+ signaling. Cell. Mol. Life Sci. 2013, 70, 2697–2712.
- Paul, B.M.; Vanden Heuvel, G.B. Kidney: Polycystic kidney disease. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 465–487.
- Salvadori, M.; Tsalouchos, A. Novel Therapeutic Strategies Targeting Molecular Pathways of Cystogenesis in Autosomal Polycystic Kidney Disease. J. Ren. Hepatic Disord. 2017, 1, 35.
- Menezes, L.F.; Germino, G.G. The pathobiology of polycystic kidney disease from a metabolic viewpoint. Nat. Rev. Nephrol. 2019, 15, 735–749.
- Eccles, M.R.; Stayner, C.A. Polycystic kidney diseas—Where gene dosage counts. F1000Prime Rep. 2014, 6, 1–6.
- Happé, H.; Peters, D.J.M. Translational research in ADPKD: Lessons from animal models. Nat. Rev. Nephrol. 2014, 10, 587–601.
- Takakura, A.; Contrino, L.; Zhou, X.; Bonventre, J.V.; Sun, Y.; Humphreys, B.D.; Zhou, J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum. Mol. Genet. 2009, 18, 2523–2531.
- Weimbs, T. Third-hit signaling in renal cyst formation. J. Am. Soc. Nephrol. 2011, 22, 793–795.
- Kurbegovic, A.; Trudel, M. Acute kidney injury induces hallmarks of polycystic kidney disease. Am. J. Physiol. Physiol. 2016, 311, F740–F751.
- Taub, M.; Chuman, L.; Saier, M.H.; Sato, G. Growth of Madin-Darby canine kidney epithelial cell (MDCK) line in hormone-supplemented, serum-free medium. Proc. Natl. Acad. Sci. USA 1979, 76, 3338–3342.
- McAteer, J.A.; Evan, A.P.; Gardner, K.D. Morphogenetic clonal growth of kidney epithelial cell line MDCK. Anat. Rec. 1987, 217, 229–239.
- Grantham, J.J.; Uchic, M.; Cragoe, E.J.; Kornhaus, J.; Grantham, J.A.; Donoso, V.; Mangoo-Karim, R.; Evan, A.; McAteer, J. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int. 1989, 35, 1379–1389.
- Sharma, M.; Reif, G.A.; Wallace, D.P. Chapter 5: In vitro cyst formation of ADPKD cells. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2019; Volume 153, pp. 93–111. ISBN 9780128170823.
- Mangoo-Karim, R.; Uchic, M.; Grant, M.; Shumate, W.A.; Calvet, J.P.; Park, C.H.; Granthamt, J.J. Renal epithelial fluid secretion and cyst growth: The role of cyclic AMP. FASEB J. 1989, 3, 2629–2632.
- Booij, T.H.; Bange, H.; Leonhard, W.N.; Yan, K.; Fokkelman, M.; Kunnen, S.J.; Dauwerse, J.G.; Qin, Y.; van de Water, B.; van Westen, G.J.P.; et al. High-Throughput Phenotypic Screening of Kinase Inhibitors to Identify Drug Targets for Polycystic Kidney Disease. SLAS Discov. Adv. Life Sci. RD 2017, 22, 974–984.
- Takasato, M.; Er, P.X.; Becroft, M.; Vanslambrouck, J.M.; Stanley, E.G.; Elefanty, A.G.; Little, M.H. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 2014, 16, 118–126.
- Morizane, R.; Bonventre, J. V Kidney Organoids: A Translational Journey. Trends Mol. Med. 2017, 23, 246–263.
- Kim, Y.K.; Nam, S.A.; Yang, C.W. Applications of kidney organoids derived from human pluripotent stem cells. Korean J. Intern. Med. 2018, 33, 649–659.
- Thatava, T.; Armstrong, A.S.; De Lamo, J.; Edukulla, R.; Khan, Y.; Sakuma, T.; Ohmine, S.; Sundsbak, J.L.; Harris, P.C.; Kudva, Y.C.; et al. Successful disease-specific induced pluripotent stem cell generation from patients with kidney transplantation. Stem Cell Res. 2011, 2, 48.
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715.
- Ashammakhi, N.; Wesseling-Perry, K.; Hasan, A.; Elkhammas, E.; Zhang, Y.S. Kidney-on-a-chip: Untapped opportunities. Kidney Int. 2018, 94, 1073–1086.
- Strange, K. Drug Discovery in Fish, Flies, and Worms. ILAR J. 2016, 57, 133–143.
- Schmitt, S.M.; Gull, M.; Brändli, A.W. Engineering Xenopus embryos for phenotypic drug discovery screening. Adv. Drug Deliv. Rev. 2014, 69–70, 225–246.
- European Commission. Directive 2010/63/EU Directive 2010/63/EU of the European Parliament and of the Council on the Protection of Animals Used for Scientific Purposes; European Commission: Brussels, Belgium, 2010.
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e4.
- Westhoff, J.H.; Giselbrecht, S.; Schmidts, M.; Schindler, S.; Beales, P.L.; Tönshoff, B.; Liebel, U.; Gehrig, J. Development of an Automated Imaging Pipeline for the Analysis of the Zebrafish Larval Kidney. PLoS ONE 2013, 8, e82137.
- Bryda, E.C. The Mighty Mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211.
- Menezes, L.F.; Germino, G.G. Murine Models of Polycystic Kidney Disease. Drug Discov. Today Dis. Mech. 2013, 10, e153–e158.
- Igarashi, P.; Somlo, S. Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2384–2398.
- Zhang, L.; Vijg, J. Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annu. Rev. Genet. 2018, 52, 397–419.
- Biller, D.S.; DiBartola, S.P.; Eaton, K.A.; Pflueger, S.; Wellman, M.L.; Radin, M.J. Inheritance of Polycystic Kidney Disease in Persian Cats. J. Hered. 1996, 87, 1–5.
- Eaton, K.A.; Biller, D.S.; DiBartola, S.P.; Radin, M.J.; Wellman, M.L. Autosomal Dominant Polycystic Kidney Disease in Persian and Persian-cross Cats. Vet. Pathol. 1997, 34, 117–126.
- Pedersen, K.M.; Pedersen, H.D.; Haggstrom, J.; Koch, J.; Ersbøll, A.K. Increased Mean Arterial Pressure and Aldosterone-to-Renin Ratio in Persian Cats with Polycystic Kidney Disease. J. Vet. Intern. Med. 2003, 17, 21–27.
- Lyons, L.A.; Biller, D.S.; Erdman, C.A.; Lipinski, M.J.; Young, A.E.; Roe, B.A.; Qin, B.; Grahn, R.A. Feline polycystic kidney disease mutation identified in PKD1. J. Am. Soc. Nephrol. 2004, 15, 2548–2555.
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Mrug, M.; Lyons, L.A.; Weimbs, T.; Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; et al. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023.
- He, J.; Li, Q.; Fang, S.; Guo, Y.; Liu, T.; Ye, J.; Yu, Z.; Zhang, R.; Zhao, Y.; Hu, X.; et al. Pkd1 mono-allelic knockout is sufficient to trigger renal cystogenesis in a mini-pig model. Int. J. Biol. Sci. 2015, 11, 361–369.
- Lian, X.; Wu, X.; Li, Z.; Zhang, Y.; Song, K.; Cai, G.; Li, Q.; Lin, S.; Chen, X.; Bai, X.Y. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br. J. Pharm. 2019, 176, 711–724.
- Gutierrez, K.; Dicks, N.; Glanzner, W.G.; Agellon, L.B.; Bordignon, V. Efficacy of the porcine species in biomedical research. Front. Genet. 2015, 6, 293.
- Ibrahim, Z.; Busch, J.; Awwad, M.; Wagner, R.; Wells, K.; Cooper, D.K.C. Selected physiologic compatibilities and incompatibilities between human and porcine organ systems. Xenotransplantation 2006, 13, 488–499.
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin Markedly Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 46–51.
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wüthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han: SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transpl. 2006, 21, 598–604.
- Wu, M.; Wahl, P.R.; Le Hir, M.; Wäckerle-Men, Y.; Wüthrich, R.P.; Serra, A.L. Everolimus Retards Cyst Growth and Preserves Kidney Function in a Rodent Model for Polycystic Kidney Disease. Kidney Blood Press. Res. 2007, 30, 253–259.
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471.
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and Kidney Growth in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 820–829.
- Walz, G.; Budde, K.; Mannaa, M.; Nürnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 830–840.
- Gattone, V.H.; Wang, X.; Harris, P.C.; Torres, V.E. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003, 9, 1323–1326.
- Torres, V.E.; Wang, X.; Qian, Q.; Somlo, S.; Harris, P.C.; Gattone, V.H. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004, 10, 363–364.
- Wang, X.; Gattone, V.; Harris, P.C.; Torres, V.E. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J. Am. Soc. Nephrol. 2005, 16, 846–851.
- Meijer, E.; Gansevoort, R.T.; De Jong, P.E.; Van Der Wal, A.M.; Leonhard, W.N.; De Krey, S.R.; Van Den Born, J.; Mulder, G.M.; Van Goor, H.; Struck, J.; et al. Therapeutic potential of vasopressin V2 receptor antagonist in a mouse model for autosomal dominant polycystic kidney disease: Optimal timing and dosing of the drug. Nephrol. Dial. Transpl. 2011, 26, 2445–2453.
- Gattone, V.H.; Maser, R.L.; Tian, C.; Rosenberg, J.M.; Branden, M.G. Developmental expression of urine concentration-associated genes and their altered expression in murine infantile-type polycystic kidney disease. Dev. Genet. 1999, 24, 309–318.
- Sullivan, J.; Karra, K.; Moxon, S.A.T.; Vallejos, A.; Motenko, H.; Wong, J.D.; Aleksic, J.; Balakrishnan, R.; Binkley, G.; Harris, T.; et al. InterMOD: Integrated data and tools for the unification of model organism research. Sci. Rep. 2013, 3, 1802.


