Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Blanca Angelica Vega Alanis | + 4002 word(s) | 4002 | 2021-11-09 09:39:11 | | | |
| 2 | Peter Tang | -34 word(s) | 3968 | 2021-11-29 03:04:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vega Alanis, B.A. Allosteric GABAA Receptor Modulators. Encyclopedia. Available online: https://encyclopedia.pub/entry/16449 (accessed on 07 February 2026).
Vega Alanis BA. Allosteric GABAA Receptor Modulators. Encyclopedia. Available at: https://encyclopedia.pub/entry/16449. Accessed February 07, 2026.
Vega Alanis, Blanca Angelica. "Allosteric GABAA Receptor Modulators" Encyclopedia, https://encyclopedia.pub/entry/16449 (accessed February 07, 2026).
Vega Alanis, B.A. (2021, November 26). Allosteric GABAA Receptor Modulators. In Encyclopedia. https://encyclopedia.pub/entry/16449
Vega Alanis, Blanca Angelica. "Allosteric GABAA Receptor Modulators." Encyclopedia. Web. 26 November, 2021.
Copy Citation
Among the mammalian ligand-gated ion channels, the GABAA receptor family comprises the largest family with subunits encoded by 19 different genes. Some of these undergo alternative splicing, and, thereby, increase the variety. Their endogenous ligand known as the gamma-aminobutyric acid (GABA) has been established as the main inhibitory neurotransmitter in the central nervous system.
GABAA modulators
allosteric modulators
heterocyclic modulators
heterocyclic synthesis
nitrogen heterocycles
1. Introduction
Heterocycles are an extremely diverse compound class since any cyclic system in which at least one carbon is exchanged for any other atom belongs to it. Consequently, they occur abundantly in nature, where they exhibit important functions. For example, the purine and pyrimidine bases in DNA, are heterocyclic, or the amino acids proline, histidine, and tryptophan contain heterocyclic motifs. Naturally, many heterocyclic scaffolds can be found in biologically active natural products such as coniine, atropine, tetrodotoxin, etc. This list could be extended ad infinitum. Hence, it is not surprising that drug development also makes excessive use of heterocyclic scaffolds. Their use enabled us to explore and expand the drug-like chemical space. Some privileged structures contain heterocyclic moieties, which are most frequently nitrogen, oxygen, and sulfur. Heterocycles are useful structures to optimize ADME/toxicity features of drug candidates, such as solubility, lipophilicity, or their hydrogen bond donor/acceptor properties. The presence of heterocyclic moieties in recently developed drugs has increased due to advances in synthetic chemistry such as the development of cross-coupling or hetero-coupling reactions, which allow synthetic accessibility to molecules containing functionalized heterocycles. Additionally, some heterocyclic drug compounds have been developed to be inspired by natural product isolates [1].
Even though many small synthetic organic molecules with high medicinal potential contain heterocyclic rings, the range of easily accessible and suitably functionalized heterocyclic building blocks is surprisingly limited, and the construction of even a small array of relevant heterocyclic compounds is far from trivial. Heterocyclic chemistry, therefore, continues to attract the attention of medicinal and synthetic chemists. The development of novel methodologies allowing for efficient access to heterocycles is still highly beneficial [2]. In particular, synthesis of heterocyclic scaffolds has to implement novel reaction technologies for expanding the chemical space of biologically-active molecules such as biocatalytic methods [3], electrochemical methods, flow chemistry [4], and photochemistry [5] to expedite medicinal chemistry programs [6][7].
1.1. The Receptor Family
Among the mammalian ligand-gated ion channels, the GABAA receptor family comprises the largest family with subunits encoded by 19 different genes. Some of these undergo alternative splicing, and, thereby, increase the variety [8][9]. Their endogenous ligand known as the gamma-aminobutyric acid (GABA) has been established as the main inhibitory neurotransmitter in the central nervous system [10]. These chloride channels are homopentamers or heteropentamers, whereby the subunits share a 30–80% sequence identity [11]. It is generally accepted (though not proven) and suggested by the bulk of experimental evidence that the majority of GABAA receptors in the adult brain are pentamers with the subunit composition two α1, two β2, or β3 and one γ2 subunits, i.e., (α1)2(β2 or β3)2(γ2)1 [9]. The subunit arrangement of this type of receptor is depicted in Figure 1. Each subunit is defined by its position in the pentamer and by its interfaces perceived from the extracellular side, going into counter-clockwise directions. A principal (or plus, +) face is in contact with the next complementary (or minus, −) face, i.e., the canonical orthosteric β+/α− interface.
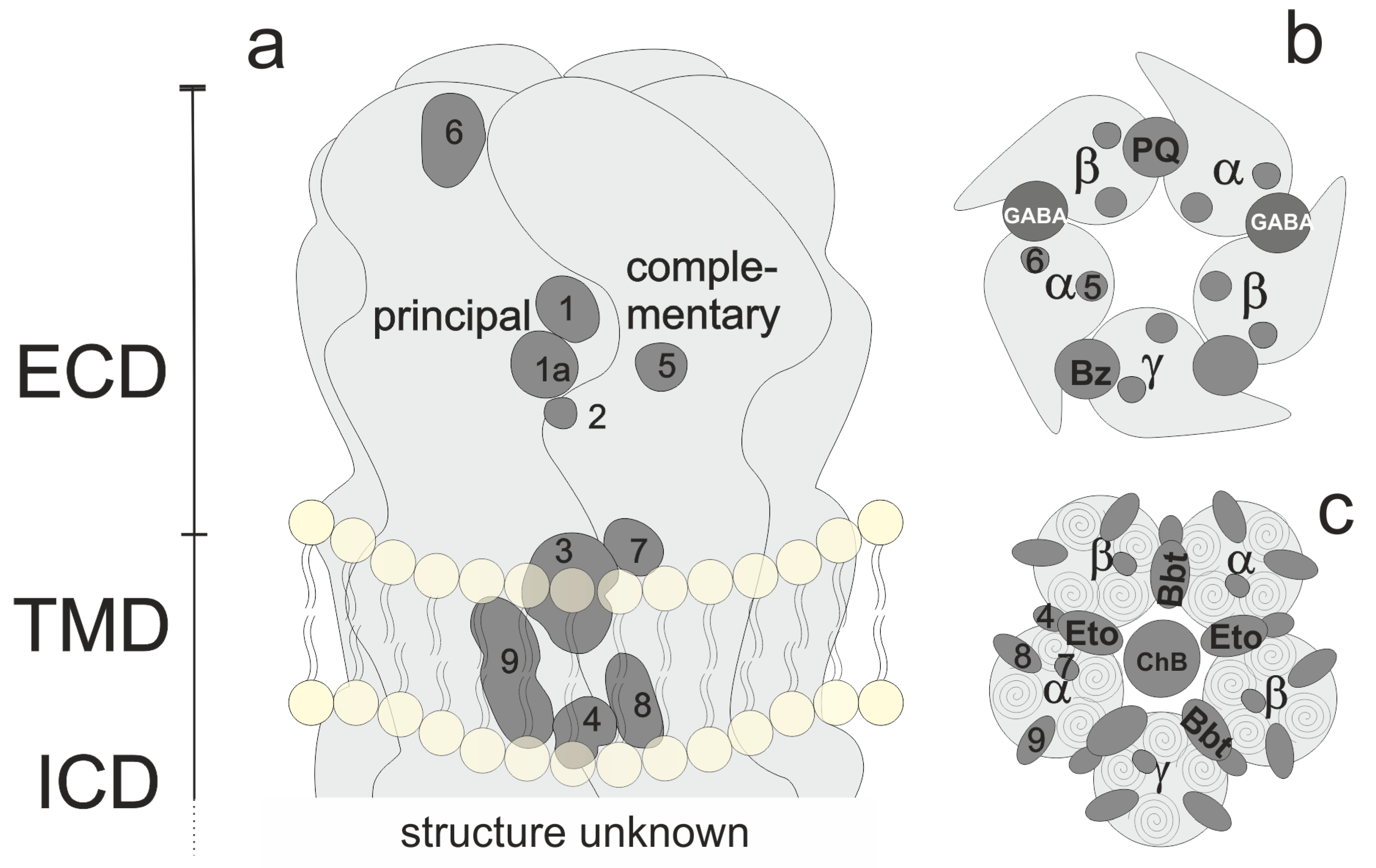
Figure 1. Overview of pentamer structure and localization of orthosteric and allosteric binding sites. (a): Generic subunit interface, featuring ECD, TMD, and lower TMD. (b) View of the ECD. GABA sites, the benzodiazepine (BZD) site, and the modulatory pyrazoloquinolinone (PQ) site are shown explicitly. (c) View of the TMD of a canonical receptor with etomidate (Eto) and barbiturate (Bbt) sites shown explicitly.
For heteromeric receptors of different compositions (such as αβ or αβδ combinations), the arrangements are debated controversially and have not yet been defined conclusively [12][13]. The number of pentameric assemblies that exist has also not been determined [9]. Figure 1 depicts a generic subunit interface, which features sites known or proposed at and near a subunit interface. Regions 1 and 1a form the extracellular domain (ECD) interface site which harbors, e.g., GABA or benzodiazepine sites (see panel b). Region 2 is a cation site while region 3 corresponds with the etomidate or barbiturate sites in the upper transmembrane domain (TMD) (see panel c), while region 4 at the lower TMD is the site where positive modulatory steroids have been observed [14]. Sites more remote to the interface (intrasubunit-sites and lipid-associated sites) for which ligands are confirmed include site 9 [14] for negative modulatory steroids, and tentative sites in positions 5, 6, 7, and 8 [15]. One α subunit shows the remaining sites with the numbers to illustrate their approximate localization from this perspective.
For the major receptor subtypes that have been observed in the mammalian central nervous system, subtype-selective targeting to address specific circuitry and physiology selectively has been attempted and is still an ongoing major direction of ligand development [16]. Knowledge of binding sites is crucial for subtype-selective targeting so that pockets that feature sequence differences can be taken into consideration as discussed for some relevant examples below.
1.2. Allosteric Binding Sites
GABAA receptors are targets of many heavily used pharmaceuticals such as benzodiazepine-based sedatives, hypnotics, and anxiolytics, barbiturate-based anticonvulsants, and general anesthetics of a broad range of chemotypes [16][17]. Nearly all pharmaceuticals and a wide range of toxins that target members of the GABAA receptor family do not interact with the binding site for the physiological agonist (GABA) as orthosteric ligands, but rather with one or several allosteric-binding sites [12][15][17]. Figure 1 provides an overview of the described allosteric sites.
Of the sites depicted in Figure 1, some display a high degree of variability, while others are fairly conserved throughout the family [15]. The extracellular interfaces have large variable pocket-forming regions and are, thus, particularly suited for the development of selective agents. The ECD α+/γ− interface features potentially 18 subtypes (six α and three γ subunit isoforms) and is the major site of action of high-affinity benzodiazepine effects [18].
Since their serendipitous discovery and the introduction of chlordiazepoxide (Librium®) in 1960, benzodiazepines have spurred enormous efforts to develop benzodiazepine site ligands with further improved properties compared to classical benzodiazepines. Dozens of chemotypes, among them β-carbolines, pyrazoloquinolinones, and imidazopyridines, have been identified as high-affinity ligands of benzodiazepine binding sites. The exact number of high and low-affinity benzodiazepine binding sites is still not completely defined, and even high affinity sites that are not localized to the canonical α+/γ− interfaces have been described [18].
Due to the high homology of the α+/β− interfaces with the α+/γ2− interfaces (see Figure 1), benzodiazepine binding site ligands such as the class of pyrazoloquinolinones can bind at both these interfaces, eliciting different functional effects [19]. While still being in an early stage, the α+/β− interfaces are considered to be highly promising targets for novel therapeutic principles as well as a wide range of tool compounds to be utilized in (human brain) imaging or research applications [20].
Some benzodiazepines also bind at non-homologous sites located in the TMD [21][22]. These sites are thought to be the main site of action of intravenous anesthetics such as etomidate or propofol. Promiscuous binding of chemotypes that bind at the high-affinity benzodiazepine binding site at both extracellular and TMD sites has been observed unexpectedly often [22].
1.3. Allosteric Ligands and Their Action on GABAA Receptors
Ligands that bind to allosteric sites can have a multitude of effects on the receptor complex in the presence or absence of orthosteric ligands. The structural basis of the GABAA receptor pharmacology has been reviewed recently based on a surge of structural data of heteropentameric receptors with various ligands present in ortho- and allosteric sites [23].
Many compounds can interact with several binding sites that are contained within a receptor. The ones that bind at the location of the endogenous ligand are known as orthosteric ligands, whereas compounds that interact in other binding sites that are different from the orthosteric binding site are known as allosteric ligands.
1.3.1. Orthosteric Ligands
Orthosteric ligands can be classified into agonists, antagonists, or inverse agonists, based on the response that their binding causes to the receptor (definitions were taken from NIH, NIC dictionary). Agonists cause the same effect as the endogenous ligand that normally binds to the receptor. Antagonists attenuate or block the effect of an agonist. Inverse agonists bind to the receptor and produce the opposite pharmacological effect that would be produced by the endogenous ligand [24].
An example of a GABAA agonist is muscimol, whereas a well-known antagonist is bicuculline [25][26][27]. Figure 2 depicts the structure of examples of an agonist and an antagonist/inverse agonist of the GABAA receptor.
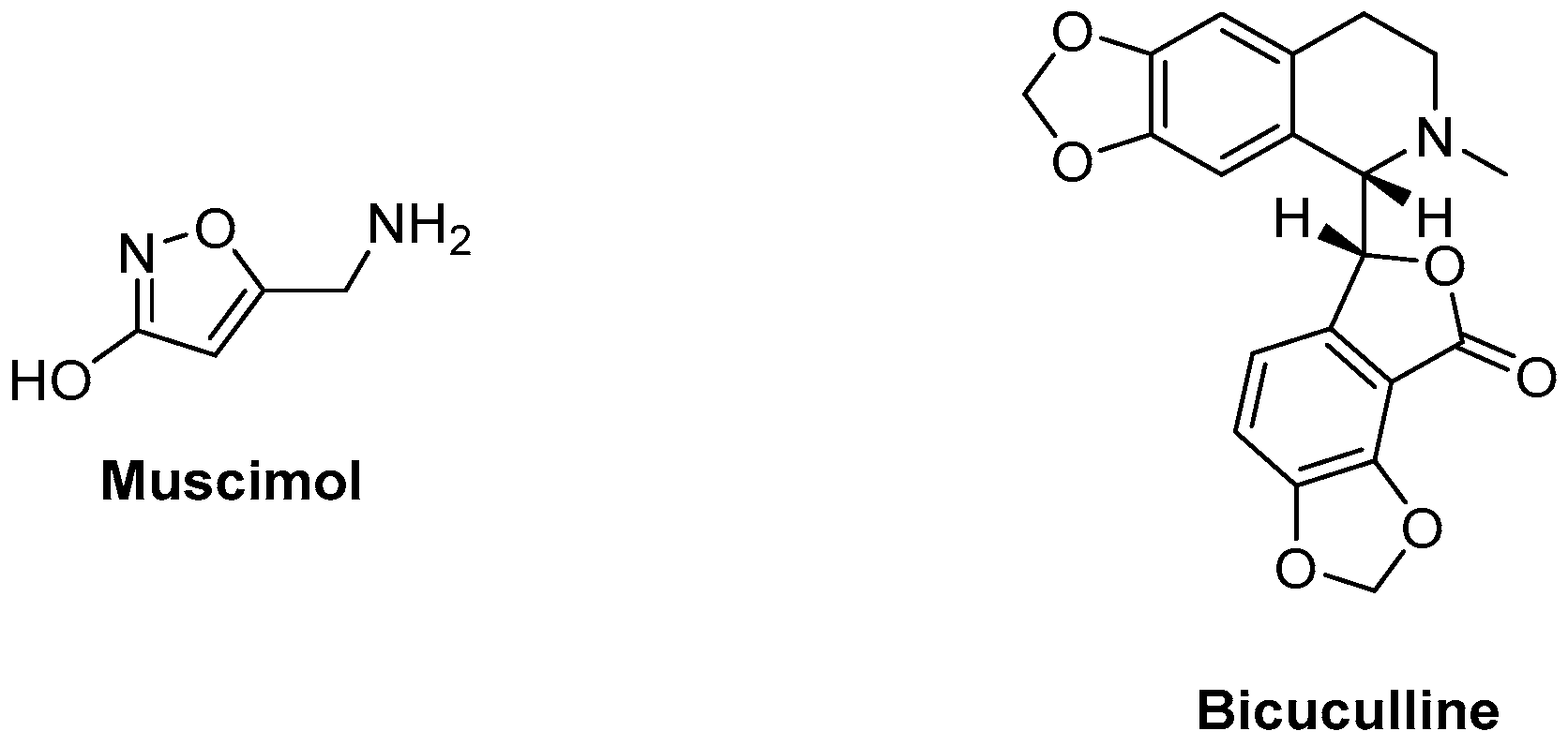
Figure 2. Examples of a GABAA receptor agonist and antagonist.
1.3.2. Allosteric Ligands
When allosteric ligands interact with a receptor, they can either exert an effect in the receptor’s physiology or just bind but produce no modification on the receptor’s physiology. In the former case, some allosteric ligands can directly exert their effects without the orthosteric ligand present. On the other hand, allosteric ligands that require the presence of the endogenous ligand to exert an effect are commonly referred to as allosteric modulators.
Allosteric ligands that produce an enhanced response when interacting with the receptor in the absence of the endogenous ligand are known as allosteric functional agonists or termed GABAmimetics. Examples of these kinds of allosteric functional agonists are etomidate, barbiturates, or neuroactive steroids [28][29]. Similarly, allosteric ligands can reduce or block channel activity in non-competitive ways. An example of a GABAA receptor blocker is picrotoxin, which has a binding site located within the channel pore [30]. Functional allosteric antagonism that is not based on a channel block can occur at a variety of allosteric sites, such as the effects elicited by inhibitory steroids [14]. Figure 3 depicts the structure of some examples of allosteric ligands, which impact the receptor function in the absence or presence of GABA.
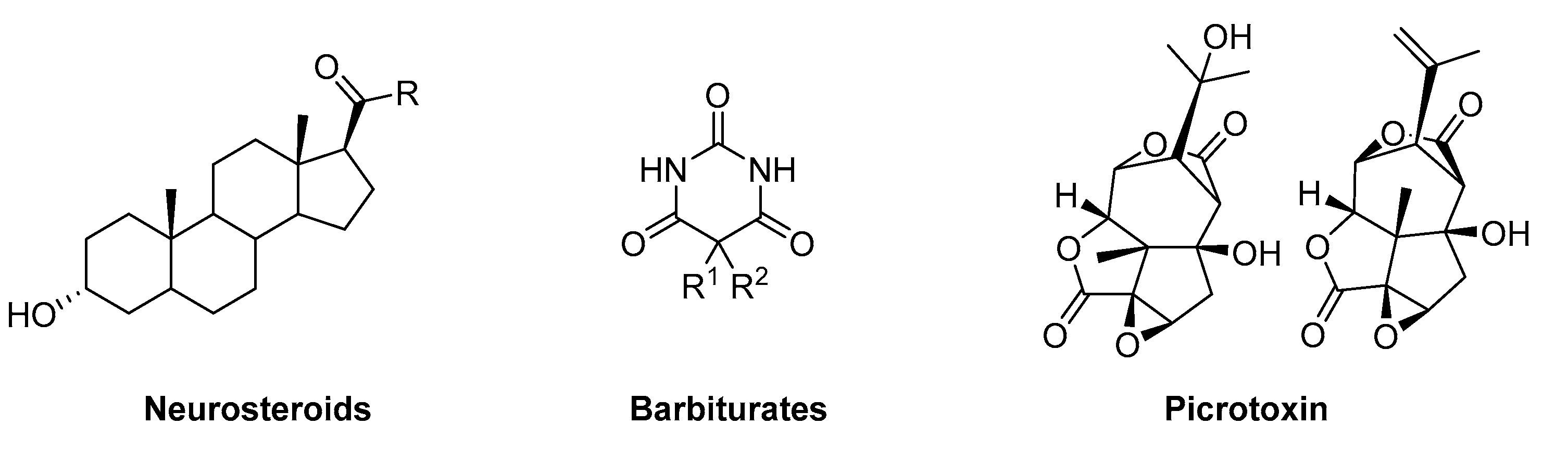
Figure 3. Examples of allosteric GABAA receptor ligands.
Allosteric modulators (in the strict sense) require the presence of the endogenous ligand to exert their effects. In other words, they modulate the GABA-elicited channel response. In general, allosteric modulators are classified in the following way.
-
Positive allosteric modulators (PAM): they enhance the effect of the endogenous ligand
-
Negative allosteric modulators (NAM): they diminish the effect of the endogenous ligand
-
Silent allosteric modulators (SAM): they do not influence the effects of the endogenous ligand, but they can compete with a PAM or a NAM for the occupation of a binding site [31].
Figure 4 shows a schematic representation of the effect that a GABAA receptor PAM, NAM, and SAM can produce in comparison to the response that GABA produces.
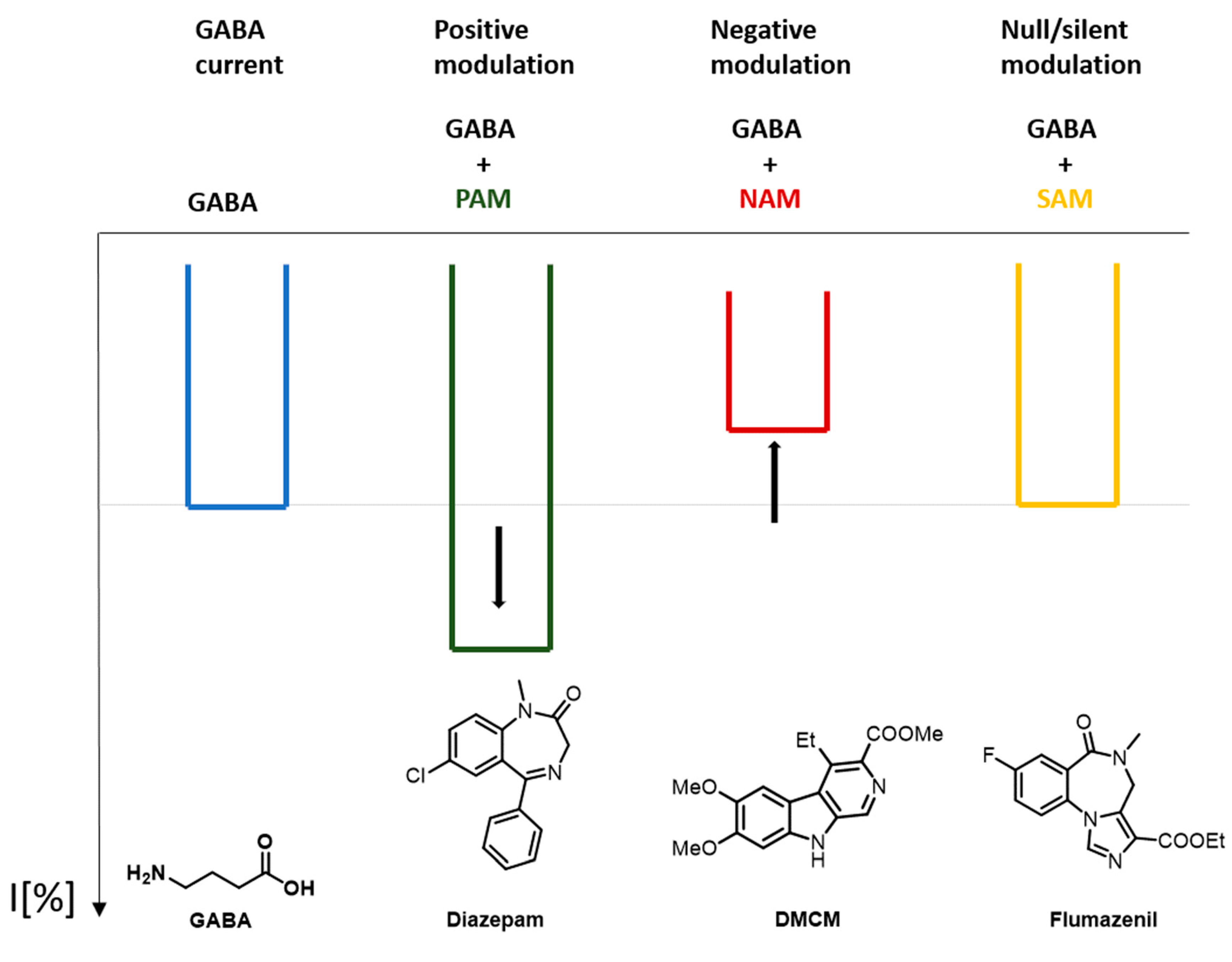
Figure 4. Effect on the response current caused by a GABAA positive allosteric modulators (PAM), negative allosteric modulators (NAM), and silent allosteric modulators (SAM).
Since some ligands can fall into more than one category of these classifications, each receptor subtype must be considered separately for each case. Some ligands can act as a SAM in a certain receptor subtype, whereas, in other receptor subtypes, it can show a PAM behavior. Flumazenil is an example of a GABAA receptor SAM [18]. Etomidate is another example of a GABAA receptor ligand with a mixed behavior. Depending on the concentration that is being studied, etomidate can behave as an allosteric modulator or as an allosteric functional agonist.
2. Assays to Probe Interactions of Small Molecules with the Receptors, or with Specific Binding Sites
Historically, radioligands were used to test substances’ ability to displace or modulate a known ligand, where usually tritiated ligands are used and where brain membranes are used as a preparation of all receptors found in the brain. To test specific receptor subtypes, membrane preparations from cells used for recombinant expression are employed (typically HEK, COS, Ltk cells, etc.). The most commonly used radioligands interact with the orthosteric site, with the high-affinity benzodiazepine sites, and with the channel pore.
Ligands of the orthosteric site (GABA sites) such as muscimol, GABA, gabazine (and likely many others) were and still are used. They display different degrees of specificity for different isoforms (subtypes). To indicate the challenges and limitations, in a recent study, it was shown that the majority of high-affinity muscimol sites in brain membranes are not the most expressed αβγ receptors, but are a smaller population of δ- containing receptors [32].
In this scenario, we are interested chiefly in allosteric ligands. Historically, radioligands for the high-affinity benzodiazepine sites predate the cloning of receptor subunits and were originally considered as ligands of an unidentified “benzodiazepine receptor.” Thus, ligands that displaced radiolabeled benzodiazepines were termed “agonists,” “antagonists,” and “inverse agonists” of the “benzodiazepine receptor.” This leads to the unfortunate situation that benzodiazepine site ligands are often referred to as “GABAA receptor agonists or inverse agonists,” which is rather misleading. Several radioligands for high-affinity benzodiazepine sites exist and are widely used: flunitrazepam, flumazenil, Ro-15 4513 are the most commonly used ones. They do not show complete overlap in the subtypes to which they are binding with high affinity. Therefore, some radioligands bind to more receptor subtypes than others due to differential interactions with isoforms of α or γ subunits.
Radioligands that bind in a state-dependent way to the ion channel also exist, where a prominent example is EBOB. EBOB binding is state-dependent. Therefore, its binding is dependent on the absence or presence of GABA and often even responds to allosteric modulators. Thus, modulation (rather than displacement) of EBOB binding can also be used to detect ligands that bind at sites other than the channel lumen, but these sites then remain unspecified [33].
Limitations of radioligand based assays include the following. Competitive displacement is the gold standard to confirm binding site usage. Radioligand modulation helps to detect ligands that use unknown sites [33], but may fail to respond to sites that cause only small or no conformational changes (silent binders that may not be silent in the presence of other agents).
As discussed in the previous chapter, ligands not only bind to the receptors, but they exert complex effects such as negative or positive modulation, or direct channel block, or allosteric “activation” on them. These effects can only be detected with functional assays. This more comprehensive approach directly assesses the effects of ligands on channel activity, or on agonist-elicited channel activity. The biggest disadvantage of functional studies is that the binding sites cannot be identified by the assay. Competitive functional assays are exceedingly challenging and often allosteric and competitive effects cannot be reliably distinguished.
The most commonly used methods to determine ligand function comprise low and high throughput electrophysiological assays and diverse methods that are based on fluorescent probes that are charge-sensitive or, otherwise, coupled to changes in membrane potential. Functional assays are performed in recombinantly expressing cell systems to determine the effects of ligands on defined receptor species or on known mixtures of subunits that are expressed, or on ex vivo preparations such as acute brain slices, synaptosome or membrane preparations, or on primary cultured cells (typically hippocampal neurons). It has to be noted that the large diversity of receptor subtypes renders every assay incomplete in the sense that effects only on those receptors that are present in a given preparation can be captured.
Due to the big complexity of GABAA receptor-mediated pharmacology and the wide diversity of assays, which are used to investigate ligand properties such as affinity, potency, electrophysiological efficacy, and in vivo effects and efficacy, it is challenging to compare ligand characteristics across different studies. Even though, by now, it is well acknowledged that radioligand-based competition assays can fail to detect modulatory interactions, there is still a lack of consensus concerning functional testing. The large number of receptor subunits, the lacking knowledge about the precise number of existing subtypes, and the observation that neurons, glia cells, and non- neuronal cells express a broad and heterogeneous mixture of subunits documents that no single in vitro or ex vivo protocol can be designed and research relies on combining multiple assay types to define ligand effects on molecular species. A recent review illustrates the complexity and discusses many of the problems that are faced on the path toward receptor subtype-specific ligand characterization [16].
Generally, after binding or functional effects are confirmed in either heterologously expressed protein or suitable ex vivo preparations (such as brain membranes, acute brain slices, or neuronal cell cultures), a broad range of toxicological and behavioral testing will follow for the most promising compounds. The behavioral animal experiments are out of the scope of this review. As a very broad rule, compounds that are chiefly GABAmimetic or positive allosteric modulators display sedative-like and anticonvulsant-like effects while ortho- and allosteric antagonists and blockers will be anxiogenic and pro-convulsant. If individual subtypes respond differently, very complex combinations of effects can occur and raise hopes for the development of totally novel therapeutics that combine silent, PAM, and NAM characteristics toward different receptor subtypes [34].
3. Clinical Pharmacology of Allosteric Ligands Used in Human Medicine
Three large groups of heavily used GABAergics are allosteric modulators of GABAA receptors, and additional indication groups exist and are summarized in this case as a fourth heterogeneous group.
3.1. Sedative General Anesthetics
Drugs with reliable sedative-hypnotic, amnestic, anxiolytic, antinociceptive, and immobilizing properties represent an indispensable component of state-of-the-art general anesthesia. After the introduction of diethyl ether as the first viable anesthetic for surgery in the middle of the 19th century, drug discovery became a prime focus of research in the field of anesthesia [35]. The ongoing quest for novel compounds with rapid-onset, potent sedative-hypnotic effects, predictable clearance to inactive metabolites and minimal side effects has yielded a range of clinically established drugs. The vast majority of anesthetic compounds in use today interact with GABAA receptors [36]. In this scenario, we provide a glance at the clinical roles of four important anesthetics, with the primary effects to rely predominantly on positive allosteric modulation of GABAA receptors: Propofol, Etomidate, Midazolam, and Thiopental (Figure 5).
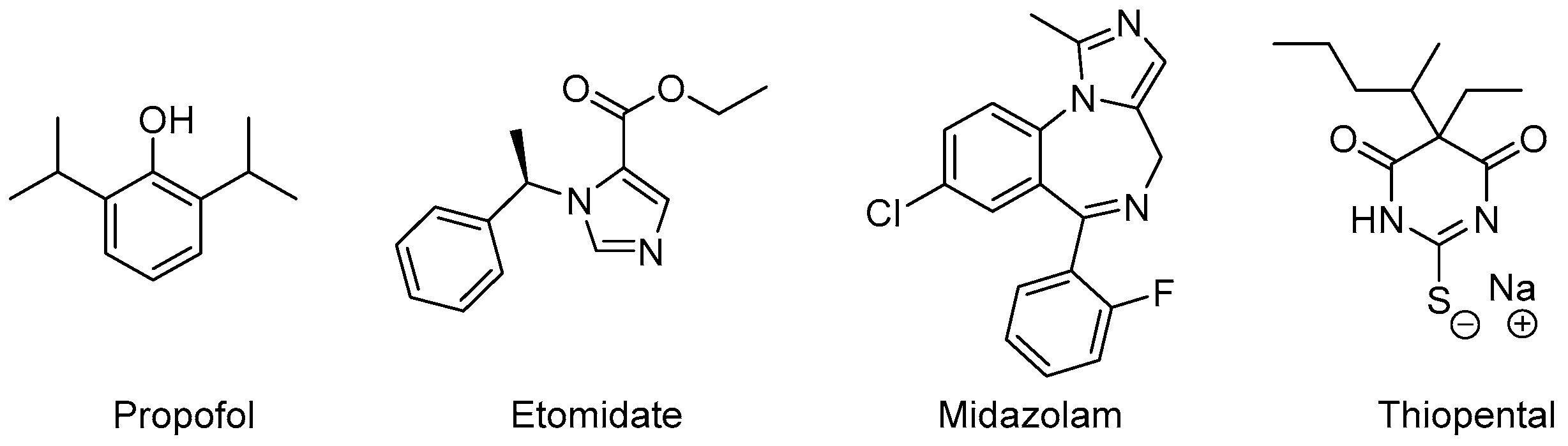
Figure 5. Anesthetic drugs with different allosteric mechanisms of action.
Propofol is a hydrophobic compound for intravenous application in a lipid-solution. It was first approved for use in the 1980s and has quickly advanced to become the most frequently administered intravenous drug for the induction of general anesthesia and a common choice for the maintenance of general anesthesia as well as procedural sedation [37]. Rapid inductions, rapid terminal half-life time, low incidence of postoperative nausea and vomiting, as well as rapid psychomotor recovery make it a very versatile hypnotic drug [38].
Propofol substantially inhibits sympathetic nerve activity, contributing to a significant, dose-dependent decrease of mean blood pressure [39] as well as moderate respiratory depression [40]. While these cardiopulmonary side-effects are predictable and well manageable in most settings, they often present a challenge in hemodynamically constrained patient populations. Other adverse events include pain on the injection site and less commonly hyperlipidemia resulting from lipid formulation.
Etomidate is an imidazole derivative, where its anesthetic activity was coincidentally discovered during animal testing as an antifungal agent in the 1960s. Its rapid onset of effects can be explained by the compound’s lipid solubility and large non-ionized fraction at physiological pH and make it a common choice for induction of general anesthesia and short procedures requiring deep sedation. The prolonged application of etomidate, however, is not feasible due to the incidence of adrenocortical suppression, which negatively impacts the outcome, especially in critically-ill populations. Compared to propofol, etomidate only causes a modest decrease of arterial blood pressure and leaves the respiratory system largely unaffected [41].
Midazolam is a short-acting, rapid-onset benzodiazepine primarily used during procedural sedation. Its low hemodynamic side-effects also make it a common choice for general anesthesia during cardiac surgery. Its amnestic effects are advantageous in treating intraoperative awareness [42] and its application may be correlated with a decreased likelihood of delayed recall of procedural sedation [43]. As with other benzodiazepines, paradoxical excitement, cognitive impairment, and prolonged recovery time present known complications.
Thiopental is an ultra-short acting barbiturate derivative. Barbiturates were used extensively over several decades to induce and maintain general anesthesia. They have been largely replaced by newer compounds with safer pharmacokinetic and pharmacodynamic profiles. Thiopental remains the only anesthetically used barbiturate with a firm place in today’s surgery rooms and emergency departments.
Many additional anesthetic compounds such as inhalational fluranes and ketamine also allosterically modulate GABAA receptors but rely on other primary mechanisms of action.
3.2. Prescription Hypnotics and Sleeping Aids
GABAA receptors present the prime molecular targets in the pharmacological treatment of insomnia. Barbiturates undoubtedly played a historically significant role as prescription hypnotics but have been largely replaced by safer options over the years. Their low therapeutic index paired with high tolerance formation and addiction potential contribute to the significant risks associated with their use [44].
Benzodiazepines are known for their ability to promote sleep, dampen anxiety, and provide muscle relaxation with a much broader therapeutic index than barbiturates. Their aggressive marketing and extensive prescription during recent decades after their market introduction are now being increasingly criticized after tremendous evidence for risks associated with long-term usage emerged. Prescription of benzodiazepines such as temazepam and nitrazepam still constitute a significant fraction of prescribed hypnotics [45]. The largest fraction of prescribed treatments for insomnia today is newer non-benzodiazepines such as zolpidem and zopiclone (Z-Drugs). While they are promoted and attributed to possessing greater benefits and fewer side effects than benzodiazepines, there is no evidence of significant differences in clinical effectiveness and safety [46].
Many nature-derived compounds of which some are used as OTC sleep-aids, also seem to mediate their sleep-promoting effects via interactions with GABAA receptors [47].
The development of new drugs with high efficacy, lower addiction liability, and lower incidence of paradoxical reactions remains imperative in order to create safer long-term treatment options for insomnia.
3.3. Anticonvulsants
Several different antiepileptic drugs with various known and unknown mechanisms of action are in use for long-term prophylactic treatment. The treatment of acute seizures and status epilepticus, however, requires potent and fast-acting drugs capable of effectively disrupting the convulsive state. Many of these compounds are modulators of GABAA receptors. Parenteral benzodiazepines such as midazolam, lorazepam, and diazepam are recommended during the initial phase of therapy, which begins after 5 min of a persistent seizure. If the seizure continues after a duration of 20 min, a second treatment phase begins, which consists of, not predominantly, GABAergic drugs such as fosphenytoin, valproic acid, or levetiracetam. Due to a higher incidence of adverse events, phenobarbital is only recommended in case the other options are unavailable. At the 40-min mark, a third treatment phase begins, where anesthetic doses of thiopental, midazolam, or propofol should be considered [48].
3.4. Additional Indication Groups for Therapeutically Used Allosteric Modulators of GABAA Receptors Exist
Other indication groups for benzodiazepines include anxiolysis and central muscle relaxation. In such cases, sedative effects are unwanted and tolerance formation limits the duration of effective treatment. Since there are differences in the effect profiles of benzodiazepines, specific derivatives are recommended for specific indications, which are not only based on their pharmacokinetic profiles [45].
Barbiturates are still regularly used in the management of severe traumatic brain injury, where lowering intracranial pressure and optimization of cerebral oxygenation are critical [49].
3.5. PET and SPECT Imaging Ligands
Allosteric ligands of specific groups of GABAA receptor subtypes are not only in use as therapeutics but also as isotope-labeled imaging ligands for positron emission tomography (PET) or single-photon computed tomography (SPECT) to investigate altered receptor expression patterns in a wide range of neuropsychiatric disorders [50]. At this time, all commonly used ligands for human brain imaging are ligands of some of the high-affinity benzodiazepine binding sites. Prominent examples are flumazenil, iomazenil, and Ro 15-4513. Imaging ligands that interact with other receptor subtypes and do not rely on the presence of a γ-subunit would be highly desired.
4. Allosteric Ligands and Their Synthetic Accessibility
Many heterocyclic scaffolds have been investigated as GABAA receptor ligands. In addition, it turns out that many of them contain nitrogen heterocycles. In fact, a large part contains even fused heterocycles, which represent scaffolds that are often complex to synthesize. Within this chapter, the more recent (last 10 years) synthetic developments in the large field of GABAA receptor ligands are summarized. This means that the selection of examples is based on new or improved synthetic methods that have been applied, and not on improved biological activity. Hence, examples synthesized via long-standing synthetic procedures are not included even though they might show interesting properties.
References
- Jampilek, J. Heterocycles in Medicinal Chemistry. Molecules 2019, 24, 3839.
- Volonterio, A.; Zanda, M. Multicomponent, One-Pot Sequential Synthesis of 1,3,5- and 1,3,5,5-Substituted Barbiturates. J. Org. Chem. 2008, 73, 7486–7497.
- Goodwin, N.C.; Morrison, J.P.; Fuerst, D.E.; Hadi, T. Biocatalysis in Medicinal Chemistry: Challenges to Access and Drivers for Adoption. ACS Med. Chem. Lett. 2019, 10, 1363–1366.
- Bogdan, A.R.; Organ, M.G. Flow Chemistry as a Drug Discovery Tool: A Medicinal Chemistry Perspective. In Flow Chemistry for the Synthesis of Heterocycles; Sharma, U.K., Van der Eycken, E.V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 319–341.
- Bogdos, M.K.; Pinard, E.; Murphy, J.A. Applications of organocatalysed visible-light photoredox reactions for medicinal chemistry. Beilstein J. Org. Chem. 2018, 14, 2035–2064.
- Boström, J.; Brown, D.G.; Young, R.J.; Keserü, G.M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 2018, 17, 709–727.
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414.
- Sieghart, W. Structure and pharmacology of gamma-aminobutyric acidA receptor subtypes. Pharmacol. Rev. 1995, 47, 181.
- Olsen, R.W.; Sieghart, W. International Union of Pharmacology. LXX. Subtypes of γ-Aminobutyric Acid(A) Receptors: Classification on the Basis of Subunit Composition, Pharmacology, and Function. Update. Pharmacol. Rev. 2008, 60, 243–260.
- Hashimoto, T. GABA receptor chloride ion channel. Nihon Rinsho. 1998, 56, 1824–1829.
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 2012, 287, 40224–40231.
- Chua, H.C.; Chebib, M. GABAA Receptors and the Diversity in their Structure and Pharmacology. Adv. Pharmacol. 2017, 79, 1–34.
- Wongsamitkul, N.; Baur, R.; Sigel, E. Toward Understanding Functional Properties and Subunit Arrangement of α4β2δ γ-Aminobutyric Acid, Type A (GABAA) Receptors. J. Boil. Chem. 2016, 291, 18474–18483.
- Laverty, D.; Thomas, P.; Field, M.; Andersen, O.J.; Gold, M.G.; Biggin, P.C.; Gielen, M.; Smart, T.G. Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat. Struct. Mol. Biol. 2017, 24, 977–985.
- Puthenkalam, R.; Hieckel, M.; Simeone, X.; Suwattanasophon, C.; Feldbauer, R.V.; Ecker, G.F.; Ernst, M. Structural Studies of GABAA Receptor Binding Sites: Which Experimental Structure Tells us What? Front. Mol. Neurosci. 2016, 9, 44.
- Sieghart, W.; Savić, M.M. International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans. Pharmacol. Rev. 2018, 70, 836.
- Sieghart, W. Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv. Pharmacol. 2015, 72, 53–96.
- Sigel, E.; Ernst, M. The Benzodiazepine Binding Sites of GABAA Receptors. Trends Pharmacol. Sci. 2018, 39, 659–671.
- Ramerstorfer, J.; Furtmuller, R.; Sarto-Jackson, I.; Varagic, Z.; Sieghart, W.; Ernst, M. The GABAA receptor alpha+beta- interface: A novel target for subtype selective drugs. J. Neurosci. 2011, 31, 870–877.
- Sieghart, W.; Ramerstorfer, J.; Sarto-Jackson, I.; Varagic, Z.; Ernst, M. A novel GABA(A) receptor pharmacology: Drugs interacting with the α(+) β(-) interface. Br. J. Pharmacol. 2012, 166, 476–485.
- Masiulis, S.; Desai, R.; Uchanski, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459.
- Iorio, M.T.; Vogel, F.D.; Koniuszewski, F.; Scholze, P.; Rehman, S.; Simeone, X.; Schnürch, M.; Mihovilovic, M.D.; Ernst, M. GABAA Receptor Ligands Often Interact with Binding Sites in the Transmembrane Domain and in the Extracellular Domain—Can the Promiscuity Code Be Cracked? Int. J. Mol. Sci. 2020, 21, 334.
- Scott, S.; Aricescu, A.R. A structural perspective on GABAA receptor pharmacology. Curr. Opin. Struct. Biol. 2019, 54, 189–197.
- NIH. NCI Dictionary of Cancer Terms. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms (accessed on 21 February 2020).
- Beaumont, K.; Chilton, W.S.; Yamamura, H.I.; Enna, S.J. Muscimol binding in rat brain: Association with synaptic GABA receptors. Brain Res. 1978, 148, 153–162.
- Andrews, P.R.; Johnston, G.A.R. Gaba agonists and antagonists. Biochem. Pharmacol. 1979, 28, 2697–2702.
- Johnston, G.A.R. Advantages of an antagonist: Bicuculline and other GABA antagonists. Br. J. Pharmacol. 2013, 169, 328–336.
- Belelli, D.; Lambert, J.J. Neurosteroids: Endogenous regulators of the GABAA receptor. Nat. Rev. Neurosci. 2005, 6, 565–575.
- Lambert, J.J.; Belelli, D.; Peden, D.R.; Vardy, A.W.; Peters, J.A. Neurosteroid modulation of GABAA receptors. Prog. Neurobiol. 2003, 71, 67–80.
- Uwai, K.; Ohashi, K.; Takaya, Y.; Ohta, T.; Tadano, T.; Kisara, K.; Shibusawa, K.; Sakakibara, R.; Oshima, Y. Exploring the Structural Basis of Neurotoxicity in C17-Polyacetylenes Isolated from Water Hemlock. J. Med. Chem. 2000, 43, 4508–4515.
- Rudolph, U.; Möhler, H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr. Opin. Pharmacol. 2006, 6, 18–23.
- Benkherouf, A.Y.; Taina, K.-R.; Meera, P.; Aalto, A.J.; Li, X.-G.; Soini, S.L.; Wallner, M.; Uusi-Oukari, M. Extrasynaptic δ-GABAA receptors are high-affinity muscimol receptors. J. Neurochem. 2019, 149, 41–53.
- Korpi, E.R.; Wong, G.; Luddens, H. Subtype specificity of gamma-aminobutyric acid type A receptor antagonism by clozapine. Naunyn Schmiedebergs Arch. Pharmacol. 1995, 352, 365–373.
- Rudolph, U.; Möhler, H. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 483–507.
- Hulsman, N.; Hollmann, M.W.; Preckel, B. Newer propofol, ketamine, and etomidate derivatives and delivery systems relevant to anesthesia practice. Best Pract. Res. Clin. Anaesthesiol. 2018, 32, 213–221.
- Antkowiak, B.; Rammes, G. GABA(A) receptor-targeted drug development -New perspectives in perioperative anesthesia. Expert Opin. Drug Discov. 2019, 14, 683–699.
- Feng, A.Y.; Kaye, A.D.; Kaye, R.J.; Belani, K.; Urman, R.D. Novel propofol derivatives and implications for anesthesia practice. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 9–15.
- Sahinovic, M.M.; Struys, M.M.R.F.; Absalom, A.R. Clinical Pharmacokinetics and Pharmacodynamics of Propofol. Clin. Pharm. 2018, 57, 1539–1558.
- Ebert, T. Sympathetic and Hemodynamic Effects of Moderate and Deep Sedation with Propofol in Humans. Anesthesiology 2005, 103, 20–24.
- Blouin, R.T.; Seifert, H.A.; Babenco, H.D.; Conard, P.F.; Gross, J.B. Propofol Depresses the Hypoxic Ventilatory Response during Conscious Sedation and Isohypercapnia. Anesthesiology 1993, 79, 1177–1182.
- Malapero, R.J.; Zaccagnino, M.P.; Brovman, E.Y.; Kaye, A.D.; Urman, R.D. Etomidate derivatives: Novel pharmaceutical agents in anesthesia. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 429–431.
- Spence, J.; Belley-Côté, E.; Devereaux, P.J.; Whitlock, R.; Um, K.; McClure, G.; Lamy, A.; LeManach, Y.; Connolly, S.; Syed, S. Benzodiazepine administration during adult cardiac surgery: A survey of current practice among Canadian anesthesiologists working in academic centres. Can. J. Anesth. 2018, 65, 263–271.
- Swann, A.; Williams, J.; Fatovich, D.M. Recall after procedural sedation in the emergency department. Emerg. Med. J. 2007, 24, 322–324.
- Schwartz, T.L.; Goradia, V. Managing insomnia: An overview of insomnia and pharmacologic treatment strategies in use and on the horizon. Drugs Context 2013, 2013, 1–10.
- Donoghue, J.; Lader, M. Usage of benzodiazepines: A review. Int. J. Psychiatry Clin. Pract. 2010, 14, 78–87.
- Dundar, Y.; Boland, A.; Strobl, J.; Dodd, S.; Haycox, A.; Bagust, A.; Bogg, J.; Dickson, R.; Walley, T. Newer hypnotic drugs for the short-term management of insomnia: A systematic review and economic evaluation. Health Technol. Assess. 2004, 8, 1–125.
- Hu, Z.; Oh, S.; Ha, T.-W.; Hong, J.-T.; Oh, K.-W. Sleep-Aids Derived from Natural Products. Biomol. Ther. 2018, 26, 343–349.
- Glauser, T.; Shinnar, S.; Gloss, D.; Alldredge, B.; Arya, R.; Bainbridge, J.; Bare, M.; Bleck, T.; Dodson, W.E.; Garrity, L.; et al. Evidence-Based Guideline: Treatment of Convulsive Status Epilepticus in Children and Adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016, 16, 48–61.
- Sheriff, F.G.; Hinson, H.E. Pathophysiology and clinical management of moderate and severe traumatic brain injury in the ICU. Semin. Neurol. 2015, 35, 42–49.
- Andersson, J.D.; Halldin, C. PET radioligands targeting the brain GABAA/benzodiazepine receptor complex. J. Label. Compd. Radiopharm. 2013, 56, 196–206.
More
Information
Subjects:
Clinical Neurology; Chemistry, Medicinal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
29 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No