


1000/1000
Hot
Most Recent
Early development of resistance to sorafenib accounts for the poor prognosis of advanced hepatocarcinoma (HCC). Autophagy, a double-edge autodegradative and recycling process, has been related to the modulation of drug sensitivity in cancer cells. The transcription factor forkhead box O3 (FOXO3) has been associated with the pathogenesis of HCC, but the involvement of FOXO3 on autophagy-related sorafenib resistance in HCC needs to be further investigated. A recent research verified that HCC cells are able to surpass sorafenib effects during chemoresistance acquisition via the upregulation of FOXO3 and the subsequent induction of a pro-survival autophagy. Hence, FOXO3-associated autophagy could constitute a novel therapeutic target in the advanced HCC landscape.
Hepatocarcinoma (HCC), one of the main tumors with the highest incidence and mortality worldwide[1], is commonly detected at advanced phases[2]. The tyrosine kinase inhibitor (TKI) sorafenib still represents the gold-standard first-line treatment against advanced HCC[3] but, unfortunately, the apparition of sorafenib-resistant tumor hepatocytes within the first six months of therapy is usual[2][3][4]. Thus, how to prevent or overcome acquisition of sorafenib resistance has become an imperative necessity in HCC.
Tumor genetic heterogeneity[5], hypoxic response[6][7] and autophagy have been related to worse tumor phenotypes and the loss of sensitivity to sorafenib[5]. Autophagy, an evolutionarily conserved catabolic process by which damaged or redundant cellular components are degraded into autophagolysosomes[8][9][10], exerts an important physiological function in the liver[9], and dysregulation of autophagy has been linked to hepatic diseases[11].
Autophagy plays a double function in sorafenib-resistant HCC, being able to encourage both sorafenib resistance or efficacy[3]. Therefore, a deeper investigation of autophagy at the molecular level must be carried out to develop novel effective autophagy-based therapies able to achieve better patient outcomes. Forkhead box O3 (FOXO3), a transcription factor belonging to the FOXO subfamily, has shown to transcriptionally regulate several autophagy-related markers[12], controlling autophagy even in tumor cells[13]. Opposite roles of FOXO3 have been found in different cancer types, acting as a tumor suppressor but also promoting tumor progression under certain conditions[14][15][16]. Besides, it has been suggested that FOXO3 could be involved in the autophagy-related modulation of drug sensitivity in tumor cells[17][18][19][20].
In spite of the potential association of FOXO3-related autophagy with sorafenib resistance, only two articles have analyzed the topic in the past[21][22]. These two previous investigations were mostly conducted under an specific hypoxic microenvironment and, surprisingly, statetements regarding how FOXO3 modulates autophagy and sorafenib sensitivity in HCC are clearly contrary[21][22]. While Liang et al.[22] reported that hypoxia activates FOXO3, inducing autophagy and sorafenib resistance; Lin et al.[21] contrarily indicated that FOXO3 overexpression under hypoxia could overcome sorafenib resistance by suppressing autophagy.
The current study aimed at unraveling the exact crosstalk between FOXO3, autophagy and sorafenib resistance in HCC using two well-established sorafenib-resistant HCC in vitro models that were independently generated from HepG2 cell line. Contrary to previous research, this investigation was fullly accomplished under normoxia, thereby unraveling sorafenib-resistant HCC cell response under oxygen availability. Moreover, HCC sample information was also analyzed to explore the role played by FOXO3 in HCC patients and its correlation with autophagic genes before conducting in vitro experiments. Furthermore, the main second-line treatment approved for sorafenib-refractory HCC —regorafenib— was employed to confirm the results and demonstrate for the first time its modulatory effects on FOXO3-mediated autophagy in sorafenib-resistant HCC.
Autophagy has been suggested to be implicated in the refractoriness to sorafenib of HCC cells[3][5][8], but its modulation by sorafenib is complex, depends on the cellular context, and remains not fully understood[2][3][5][8][23]. In this study, results from acridine orange staining and colocalization between the autophagosome marker microtubule associated protein 1 light chain 3 (LC3) and the lysosome marker lysosomal associated membrane protein 2 (LAMP2), sorafenib-resistant cells display an enhanced basal autophagy (Figure 1a,b,c). This result is supported by the higher protein levels observed in the autophagy markers UV radiation resistance-associated (UVRAG), Beclin-1 and autophagy-related 5 (Atg5) (Figure 1d). Sequestosome 1 (p62) and LC3-II levels in the sorafenib-resistant lines were also more pronounced compared to those observed in the parental sorafenib-sensitive HepG2 (Figure 1d). However, autophagy induction is traditionally characterized by increased LC3-II levels accompanied by decreased expression of p62, which is a substrate of autophagic degradation[24][25]. After exploring the source of such p62 upregulation, it was found that the p62 transcription factor nuclear factor erythroid 2-like 2 (NRF2) is also upregulated in the chemoresistant cells due to an increment in the production of reactive oxygen species (ROS), which ultimately elevates p62 expression despite being degraded via autophagy (Figure 1e–g).
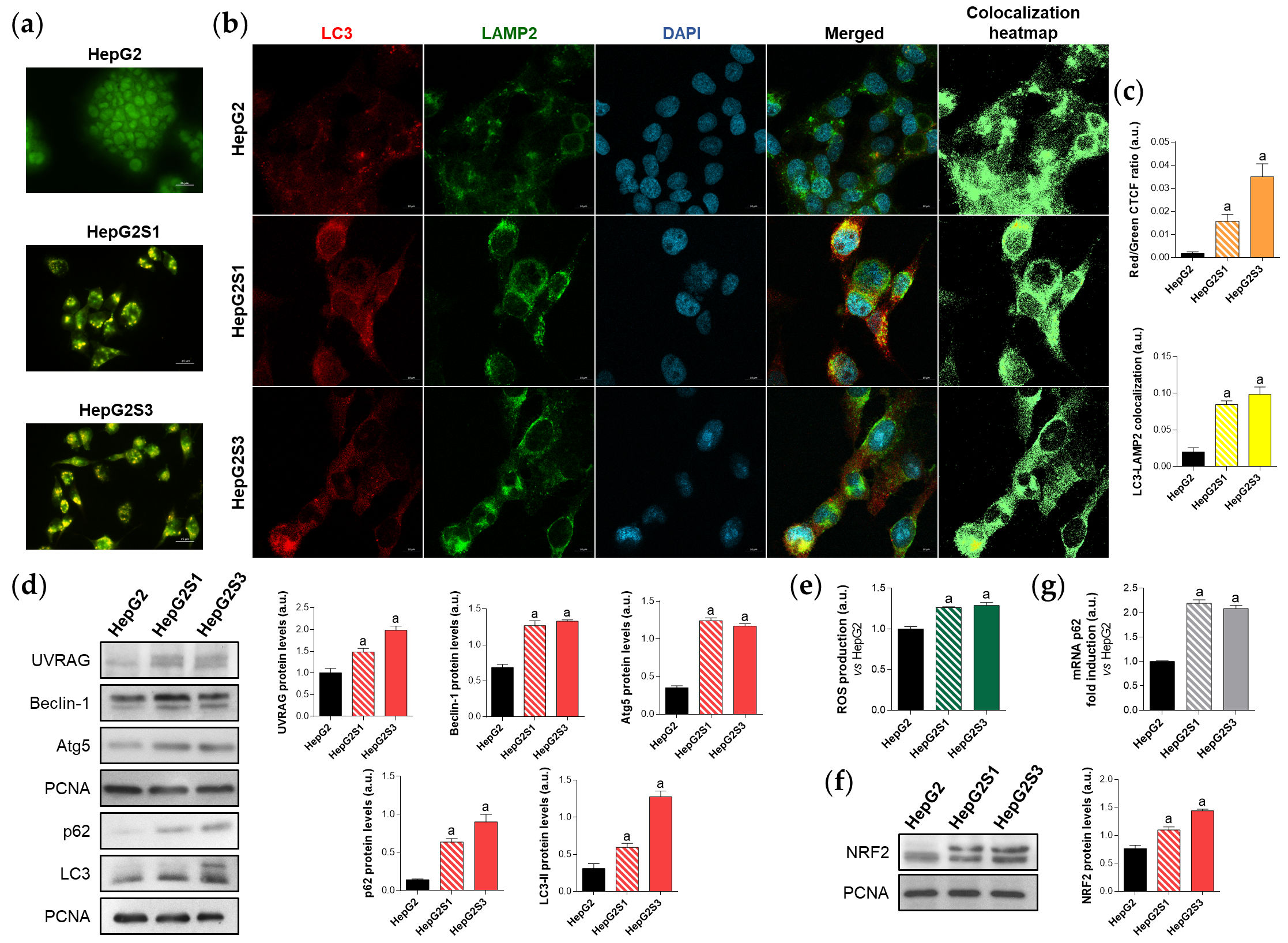
Figure 1. Comparison of basal autophagic status between HepG2S1 and HepG2S3 sorafenib-resistant lines and sorafenib-sensitive HepG2 parental cells. (a) Fluorescence microscopy images from acridine orange staining. Magnification 40×, scale bar 25 µm; (b) Confocal microscopy images from LC3-LAMP2 immunofluorescence. Yellow fluorescence in merged channel indicates LC3-LAMP2 colocalization. Magnification 63×, scale bar 10 µm. Corresponding LC3-LAMP2 colocalization heatmaps obtained using ImageJ are also shown; (c) Quantification of red/green CTCF ratio from (a) and LC3-LAMP2 colocalization from (b); (d) Representative immunoblots showing protein expression of several autophagy markers. Proliferating cell nuclear antigen (PCNA) was used as the loading control[6]. Densitometry reading of each band is shown; (e) Measurement of ROS production; (f) Immunoblots showing NRF2 protein levels. Densitometry reading of each band is shown; (g) Assessment of p62 mRNA levels. Data from (c–g) are expressed as mean values of arbitrary units (a.u.) ± SD. a p < 0.05 vs. HepG2 cells.
The abolition of the active autophagy in the sorafenib-resistant HepG2S1 and HepG2S3 lines using bafilomycin A1 or chloroquine, two recognized inhibitors of autophagosome-lysosome fusion[24], reduced the autophagolysosome amount (Figure 2a,b,c). Furthermore, treatment with such agents further enhanced p62 and LC3-II protein expression with respect to the corresponding steady-state levels (Figure 2d), indicating autophagic degradation impairment and autophagosomes accumulation. These findings highlight that the sorafenib-resistant cells are autophagy-competent and that autophagy was definitely induced in response to long-term sorafenib administration.
Additionally, autophagy suppression significantly raised the expression of the apoptotic markers Bcl-2 associated X apoptosis regulator (Bax) and cleaved caspase-3 in the resistant lines (Figure 2e), also increasing the subG1 population (Figure 2f,g) and decreasing the cell viability (Figure 2h). Altogether, these results demonstrate that chemoresistant HCC cells induced a cytoprotective autophagy to overcome the anti-tumor activity of sorafenib during the development of drug resistance.
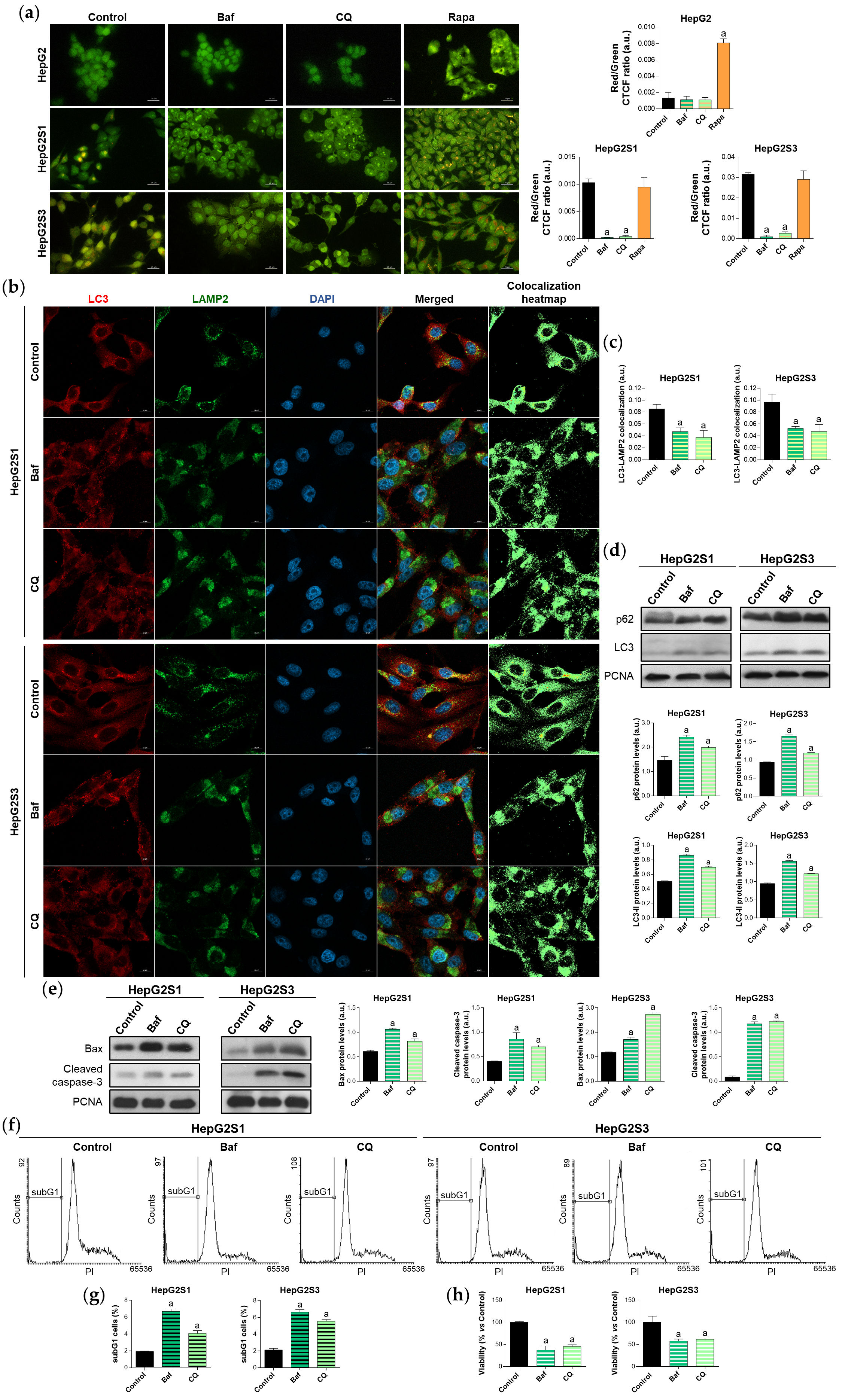
Figure 2. Effect of autophagic flux modulation on basal autophagy, cell death, and viability. HCC cell lines were treated with 100 nM bafilomycin A1 (Baf), 40 µM chloroquine (CQ) or 200 nM rapamycin (Rapa) for 24 h. (a) Fluorescence microscopy images from acridine orange staining. Magnification 40×, scale bar 25 µm. Bar graphs representing the quantification of red/green CTCF ratio are also shown; (b) Confocal microscopy images from LC3-LAMP2 immunofluorescence. Yellow fluorescence in merged channel indicates LC3-LAMP2 colocalization. Magnification 63×, scale bar 10 µm. Corresponding LC3-LAMP2 colocalization heatmaps obtained using ImageJ are also shown; (c) Bar graphs representing the quantification of LC3-LAMP2 colocalization from (b); (d) Immunoblots showing p62 and LC3 turnover. Densitometry reading of each band is shown; (e) Immunoblots showing protein expression of Bax and cleaved caspase-3. Densitometry reading of each band is shown; (f,g) Analysis of subG1 cell population; (h) Evaluation of cell viability. Data from (a), (c–e) are expressed as mean values of arbitrary units (a.u.) ± SD. Data from (g,h) are expressed as the percentage of mean values ± SD. a p < 0.05 vs. control (non-treated) cells.
The underlying molecular mechanisms of autophagy-related sorafenib resistance are still unclear. It has been suggested that deregulation of FOXO3 expression or activity could provoke tumor development and boost the apparition of more aggressive tumor phenotypes such as those with chemoresistance[13][21][22][28]. Hence, FOXO3 could relate HCC sorafenib resistance and the overactivation of pro-survival autophagy.
Employing the TCGA gene analysis tool from the UALCAN database, it has been proved that FOXO3 is significantly greater expressed in tumor samples compared to normal liver tissues (Figure 3a), and that the overexpression of FOXO3 in HCC patients is associated with lower survival rates (Figure 3b). Thus, FOXO3 high expression seems to constitute a hallmark of negative prognosis in HCC.
It is known that FOXO3 is able to transcriptionally upregulate several autophagy markers, which can lead to autophagy activation[12]. Curiously, transcriptional targets of FOXO3 such as PIK3C3, ULK1, ATG12, BECN1 and ATG4B predominantly showed a moderate degree of positive correlation with FOXO3 in HCC patient samples (Table 1). In addition, expression of multiple other genes found in the autophagy KEGG pathway (hsa04140) were shown to be positively correlated with FOXO3 in HCC patients (Table 1); altogether supporting an enhanced autophagic status. These findings suggest that enhanced FOXO3 levels are not only associated with poor patient outcomes, but also with a pro-autophagic environment in HCC.
In concordance with HCC sample data where FOXO3 seemed to be linked to worse phenotypes, sorafenib-resistant HCC in vitro models, which resemble a more advanced disease stage, showed FOXO3 overexpression (Figure 3c) and greater nuclear translocation (Figure 3d), where this factor exerts its transcriptional activity.
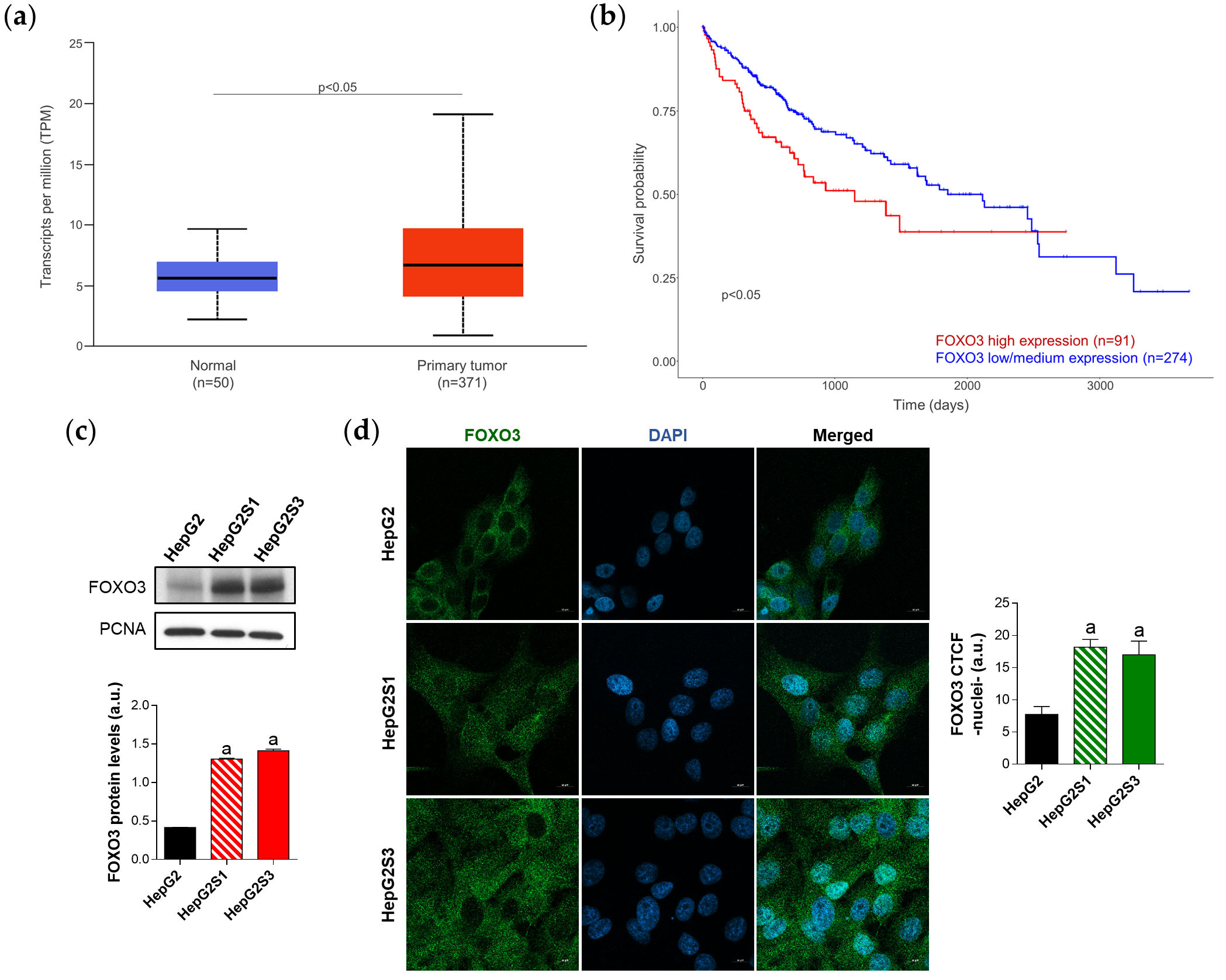
Figure 3. Characterization of FOXO3 expression in HCC patient samples and sorafenib-resistant HCC in vitro models. (a) Comparison of FOXO3 expression between HCC and normal liver tissues. p < 0.05 significant differences; (b) Impact of FOXO3 expression on HCC patient survival rate. p < 0.05 significant differences between high and low/medium FOXO3 levels; (c) Immunoblots from the in vitro analysis of FOXO3 expression. Densitometry reading of each band is shown; (d) Evaluation of FOXO3 nuclear translocation employing confocal microscopy and FOXO3 immunofluorescence. Magnification 63×, scale bar 10 µm. Bar graph from the quantification of nuclear green fluorescence (FOXO3) is also shown. Data from (c,d) are expressed as mean values of arbitrary units (a.u.) ± SD. a p < 0.05 vs. HepG2 cells.
Table 1. Autophagy-related genes positively and significantly correlated with FOXO3 in human HCC samples.
|
Gene Symbol |
Full Name |
Pearson-CC |
|
ATG5 |
Autophagy-related 5 |
+0.62 |
|
RAB33B |
RAB33B, member RAS oncogene family |
+0.6 |
|
STX17 |
Syntaxin 17 |
+0.56 |
|
ATG2B |
Autophagy-related 2B |
+0.55 |
|
SMCR8 |
SMCR8-C9orf72 complex subunit |
+0.55 |
|
TRAF6 |
TNF receptor associated factor 6 |
+0.55 |
|
UVRAG |
UV radiation resistance associated |
+0.55 |
|
PIK3C3 |
Phosphatidylinositol 3-kinase catalytic subunit type 3 |
+0.51 |
|
PIK3R4 |
Phosphoinositide-3-kinase regulatory subunit 4 |
+0.51 |
|
TANK |
TRAF family member associated NFKB activator |
+0.51 |
|
ATG16L1 |
Autophagy-related 16 like 1 |
+0.48 |
|
HMGB1 |
High mobility group box 1 |
+0.48 |
|
AMBRA1 |
Autophagy and beclin-1 regulator 1 |
+0.47 |
|
TBK1 |
TANK binding kinase 1 |
+0.47 |
|
RAB1A |
RAB1A, member RAS oncogene family |
+0.44 |
|
RAB7A |
RAB7A, member RAS oncogene family |
+0.42 |
|
ULK1 |
Unc-51 like autophagy activating kinase 1 |
+0.41 |
|
SH3GLB1 |
SH3 domain containing GRB2 like, endophilin B1 |
+0.4 |
|
ATG16L2 |
Autophagy-related 16 like 2 |
+0.38 |
|
ATG4C |
Autophagy-related 4C cysteine peptidase |
+0.38 |
|
ATG9A |
Autophagy-related 9A |
+0.38 |
|
ATG12 |
Autophagy-related 12 |
+0.37 |
|
NRBF2 |
Nuclear receptor binding factor 2 |
+0.37 |
|
SNAP29 |
Synaptosome associated protein 29 |
+0.37 |
|
C9orf72 |
C9orf72-SMCR8 complex subunit |
+0.36 |
|
DAPK1 |
Death associated protein kinase 1 |
+0.36 |
|
ATG3 |
Autophagy-related 3 |
+0.35 |
|
WDR41 |
WD repeat domain 41 |
+0.35 |
|
RB1CC1 |
RB1 inducible coiled-coil 1 |
+0.34 |
|
BECN1 |
Beclin-1 |
+0.31 |
|
ATG4B |
Autophagy-related 4B cysteine peptidase |
+0.3 |
Pearson-CC, Pearson-correlation coefficient.
There is evidence supporting that FOXO3 deregulation is associated with autophagy modulation, being related to the loss or gain of chemosensitivity in cancer[17][18][19][20][29]. Nonetheless, the relationship between sorafenib response, autophagy and FOXO3 in HCC remains largely unknown.
To assess whether FOXO3 upregulation accounts for the induction of autophagy in sorafenib-resistant HCC cells, FOXO3 was transiently silenced, reaching a successful silencing efficiency (Figure 4a) and also significantly reducing FOXO3 nuclear retention (Figure 4b). Moreover, expression of the autophagy targets ULK1, Beclin-1 and LC3 was decreased after FOXO3 knockdown (Figure 4a), and the sorafenib-mediated induction of autophagic flux was suppressed (Figure 4c,d). Therefore, the activation of autophagy caused by sorafenib sustained treatment of HCC cells is mediated, at least in part, by FOXO3 upregulation.
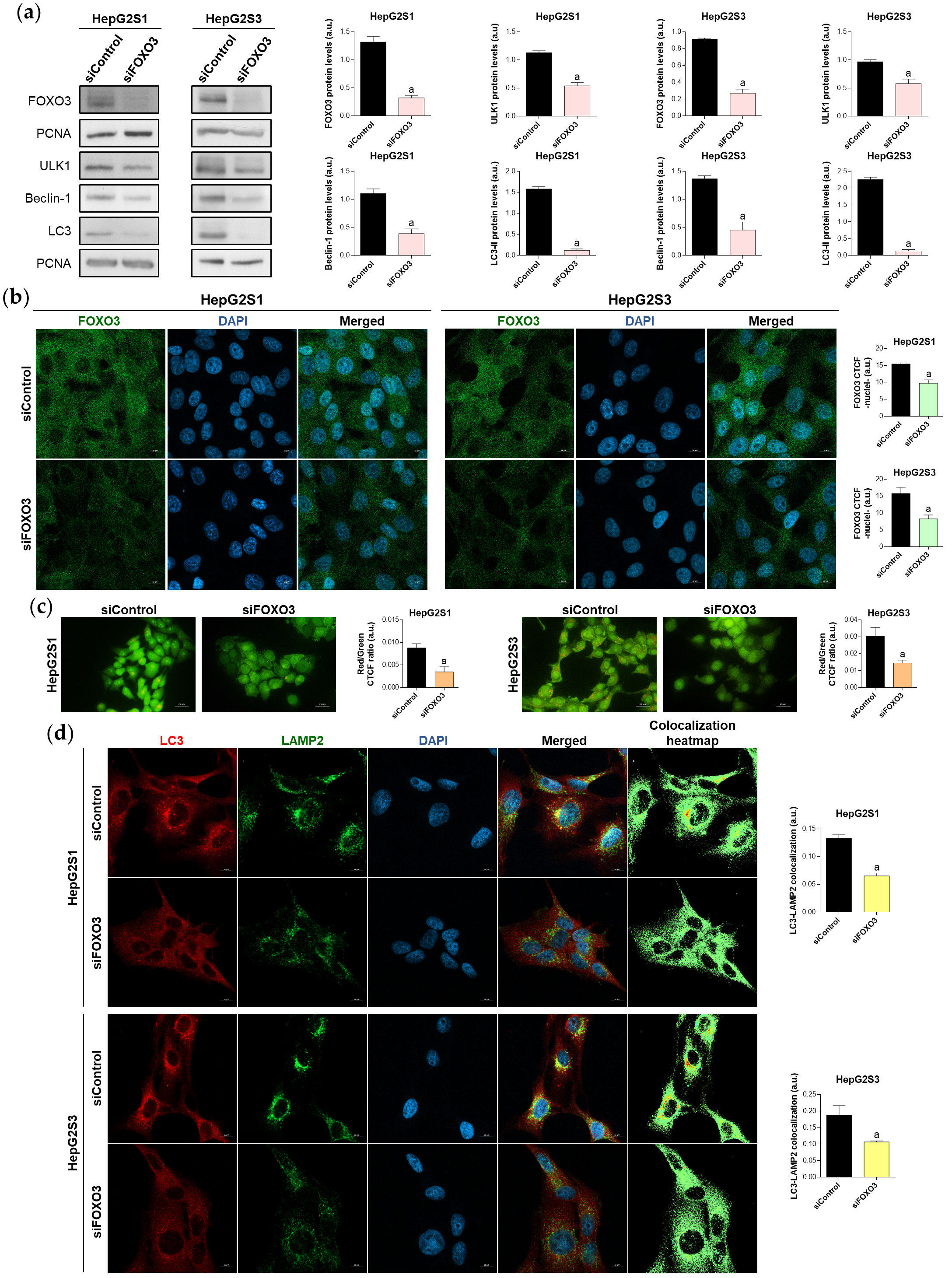
Figure 4. Effect of FOXO3 knockdown on the basal autophagic status of HepG2S1 and HepG2S3 sorafenib-resistant cell lines. All the assays were carried out at 48 h post-silencing. (a) Immunoblots of FOXO3 and several autophagy targets. Densitometry reading of each band is shown; (b) Analysis of FOXO3 nuclear translocation by confocal microscopy and FOXO3 immunofluorescence. Magnification 63×, scale bar 10 µm. Quantification of nuclear green fluorescence (FOXO3) is also shown; (c) Fluorescence microscopy images from acridine orange staining. Magnification 40×, scale bar 25 µm. Bar graphs representing the quantification of red/green CTCF ratio are also shown; (d) Confocal microscopy images from LC3-LAMP2 immunofluorescence. Yellow fluorescence in merged channel indicates LC3-LAMP2 colocalization. Magnification 63×, scale bar 10 µm. Corresponding LC3-LAMP2 colocalization heatmaps obtained using ImageJ and bar graphs representing the quantification of LC3-LAMP2 colocalization are also shown. Data from (a–d) are expressed as mean values of arbitrary units (a.u.) ± SD. a p < 0.05 vs. siControl cells.
Once verified that FOXO3 is responsible for the induction of autophagy in the chemoresistant lines, it was determined the impact of FOXO3 silencing in the survival ability of sorafenib-resistant cells, finding that FOXO3 knockdown markedly diminished cell proliferation (Figure 5a) and viability (Figure 5b), and boosted apoptosis (Figure 5c). These results suggest that FOXO3 upregulation during the development of sorafenib resistance protects chemoresistant hepatocytes from the anti-tumor effects of this TKI.
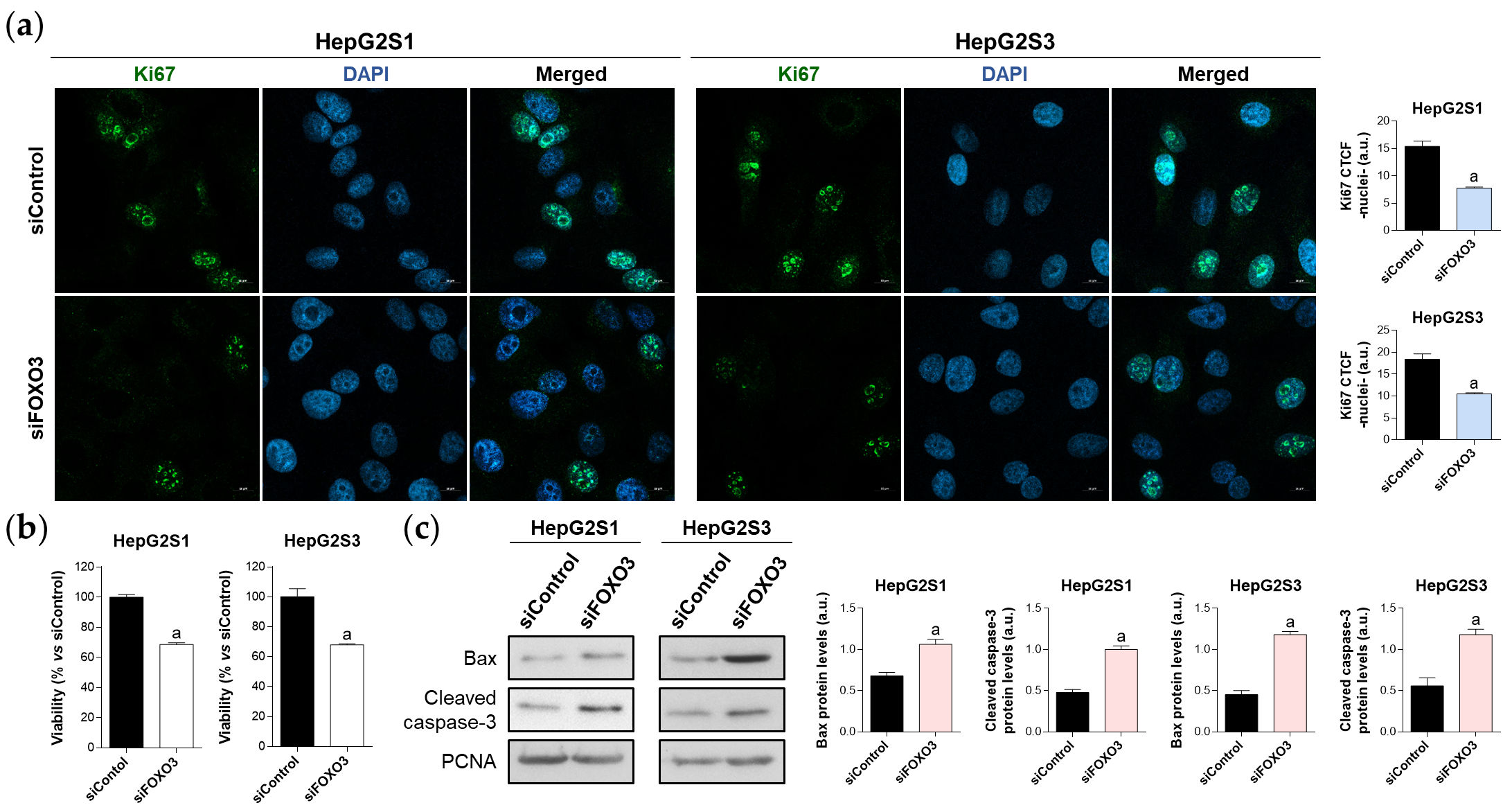
Figure 5. Effect of FOXO3 knockdown on HepG2S1 and HepG2S3 cell proliferation, viability, and apoptosis. All the assays were carried out at 48 h post-silencing. (a) Assessment of cell proliferation using Ki67 immunofluorescence and confocal microscopy. Magnification 63×, scale bar 10 µm. Quantification of nuclear green fluorescence (Ki67) is also shown; (b) Determination of cell viability; (c) Immunoblots showing Bax and cleaved caspase-3 protein levels. Densitometry reading of each band is shown. Data from (a,c) are expressed as mean values of arbitrary units (a.u.) ± SD. Data from (b) are expressed as the percentage of mean values ± SD. a p < 0.05 vs. siControl cells.
Regorafenib represents one of the main second-line treatments employed when sorafenib sensitivity is lost[30]. Although regorafenib could modulate the autophagy to counteract tumor cell survival[31], the effect of this TKI on FOXO3 and autophagy in sorafenib-resistant HCC has not been evaluated yet.
First, to assess the efficacy of regorafenib in sorafenib-resistant HCC, both cell growth and viability were analyzed after treating with a subset of regorafenib concentrations. Crystal violet experiments revealed that the inhibition of cellular growth by regorafenib is concentration- and time-dependent (Figure 6a), while cell viability at 48 h post-treatment also decreased in a dose-dependent trend (Figure 6b). Likewise, Ki67-based proliferation index was reduced by half in sorafenib-resistant cells after 48 h-exposition to 20 µM regorafenib (Figure 6c), the concentration that was established as ~IC50.
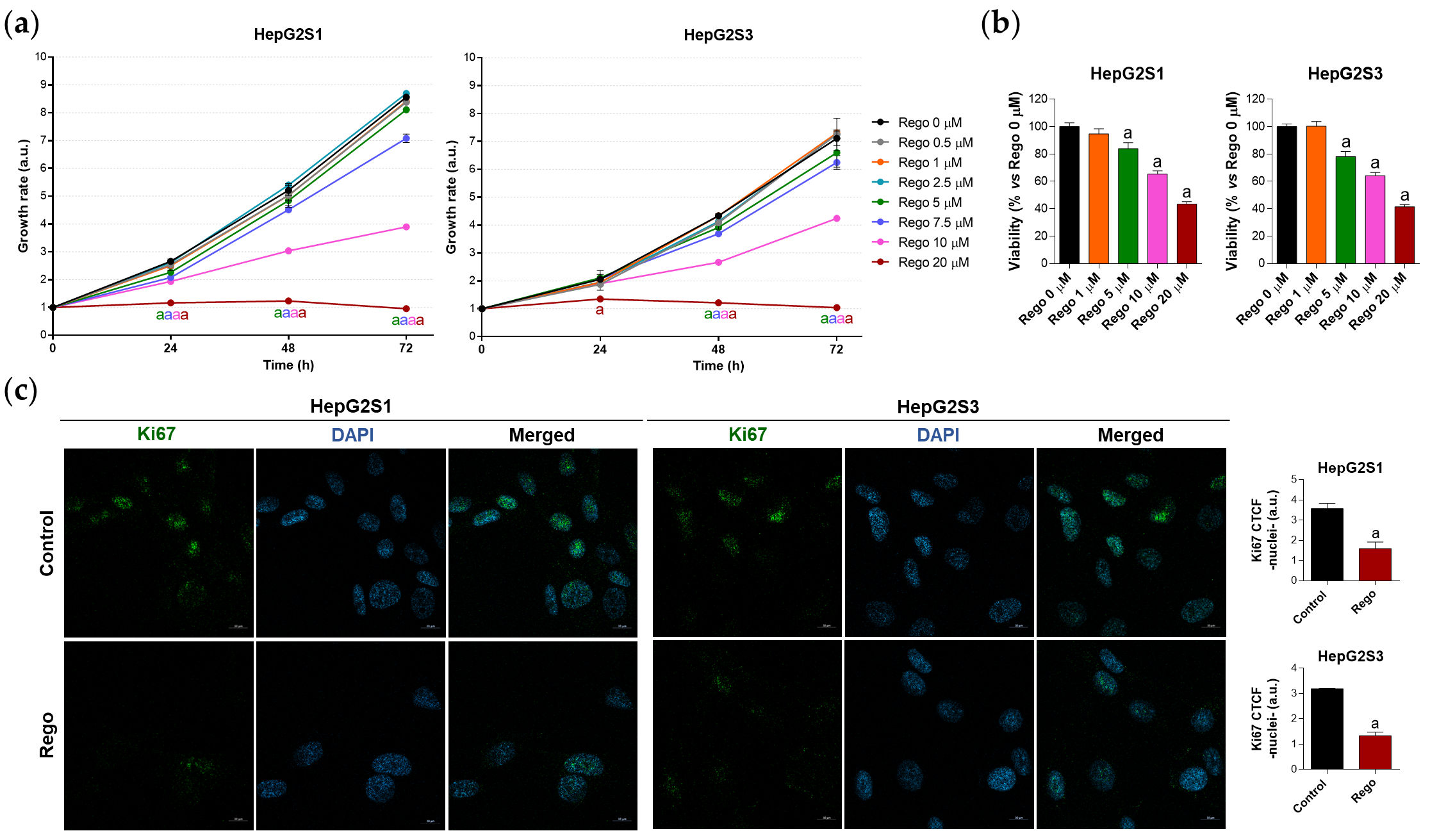
Figure 6. Evaluation of HepG2S1 and HepG2S3 cell growth, viability, and proliferation after treatment with regorafenib. (a) Determination of cell growth under several concentrations of regorafenib (Rego) using crystal violet staining. a p < 0.05 significant differences between each dosage and Rego 0 µM at each timepoint; (b) Analysis of cell viability at 48 h post-treatment with diverse concentrations of regorafenib using MTT assay. a p < 0.05 vs. Rego 0 µM; (c) Assessment of proliferation rate by Ki67 immunofluorescence and confocal microscopy after treatment with 20 µM regorafenib for 48 h. Magnification 63×, scale bar 10 µm. Quantification of nuclear green fluorescence (Ki67) is also shown. a p < 0.05 vs. control (non-treated) cells. Data from (a,c) are expressed as mean values of arbitrary units (a.u.) ± SD. Data from (b) are expressed as the percentage of the mean values ± SD.
Meanwhile, regorafenib treatment significantly downregulated FOXO3, ULK1, UVRAG, Beclin-1, and Atg5 (Figure 7a). Furthermore, nuclear retention of FOXO3 was remarkably inhibited by regorafenib (Figure 7b), which also suppressed autophagolysosome formation (Figure 7c,d). Moreover, regorafenib elevated p62 and LC3-II protein expression (Figure 7a). This surprising finding was due to the additional capability to block late autophagy through the inhibition of the fusion of autophagosomes with lysosomes (Figure 7e), which explains the effective abolishment of FOXO3-related autophagy and the simultaneous high LC3-II and p62 protein levels.
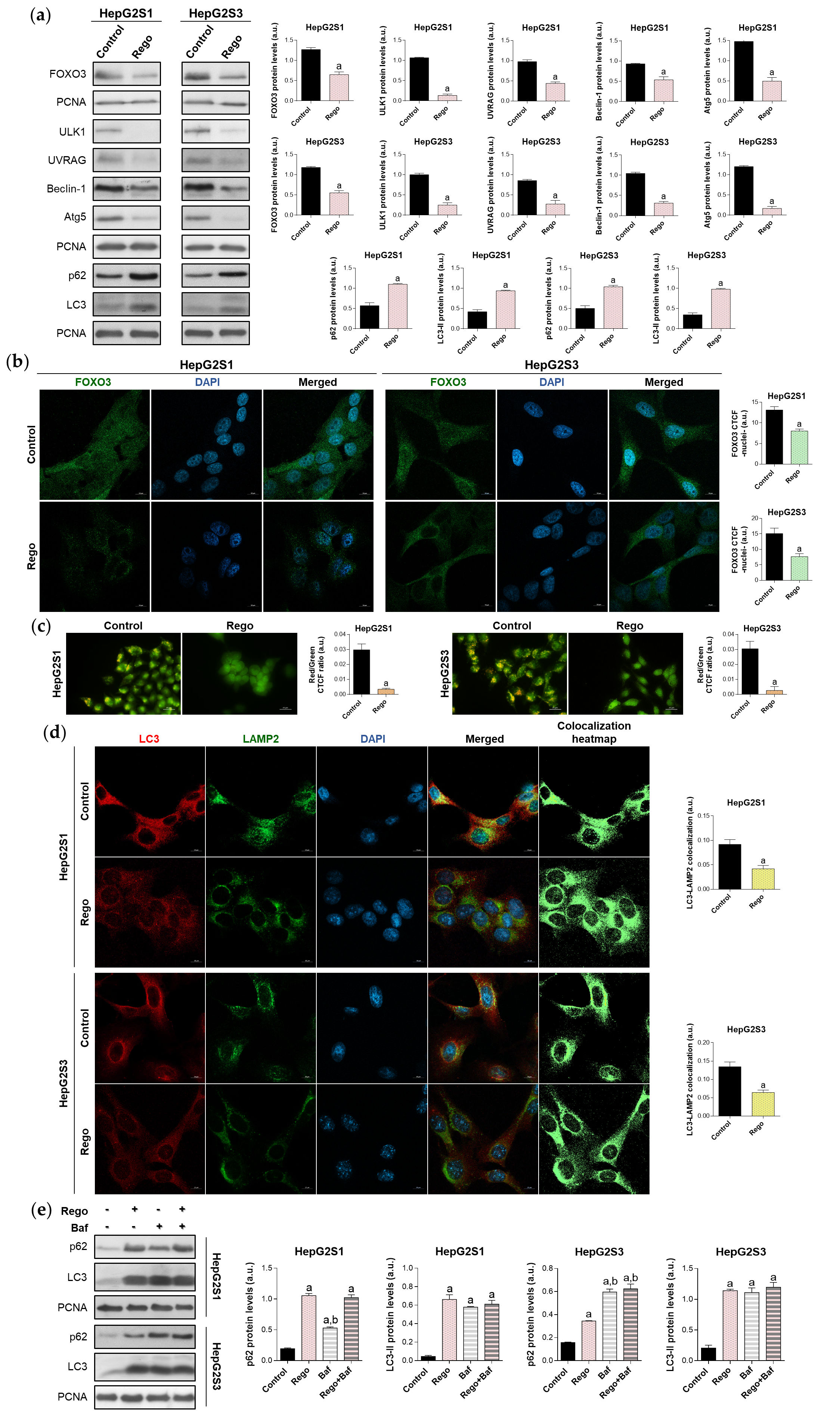
Figure 7. Impact of regorafenib treatment on the induced autophagy of HepG2S1 and HepG2S3 sorafenib-resistant cell lines. HCC cells were treated with 20 µM regorafenib (Rego) for 48 h. (a) Immunoblots of FOXO3 and autophagy-related markers. Densitometry reading of each band is shown; (b) Assessment of FOXO3 nuclear translocation by confocal microscopy and FOXO3 immunofluorescence. Magnification 63×, scale bar 10 µm. Quantification of nuclear green fluorescence (FOXO3) is also shown; (c) Fluorescence microscopy images from acridine orange staining. Magnification 40×, scale bar 25 µm. Bar graphs representing the quantification of red/green CTCF ratio are also shown; (d) Confocal microscopy images from LC3-LAMP2 immunofluorescence. Yellow fluorescence in merged channel shows LC3-LAMP2 colocalization. Magnification 63×, scale bar 10 µm. Corresponding LC3-LAMP2 colocalization heatmaps obtained using ImageJ and bar graphs representing the quantification of LC3-LAMP2 colocalization are also shown; (e) Immunoblots showing p62 and LC3 turnover after single or combined treatment with regorafenib and 100 nM bafilomycin A1 (Baf). In the case of bafilomycin A1 single administration, cells were exposed to this agent for 24 h while, for the co-treatment, cells were subjected to regorafenib for 48 h in presence of bafilomycin A1 during the last 24 h. Densitometry reading of each band is shown. Data from (a–e) are expressed as mean values of arbitrary units (a.u.) ± SD. a p < 0.05 vs. control (non-treated) cells, b p < 0.05 vs. regorafenib-treated cells.
Autophagy has been widely related to the modulation of drug efficacy in cancer, but this self-digestive mechanism can either induce drug resistance or sensitivity, showing a dual function[32]. Opposite roles of autophagy have been found in HCC under treatment with diverse chemotherapeutic agents, denoting the double role and context-dependency of autophagy[33][34][35][36][37].
The involvement of autophagy in the acquisition of resistance to sorafenib remains largely unknown. Here, this study evidenced that sorafenib-resistant HCC cells exhibit an enhanced basal autophagic flux. Accordingly, autophagy has shown to be induced in sorafenib-refractory Hep3B[38][39], HepG2[40] or Huh7[39][40][41][42] cells. Thus, sustained sorafenib administration seems to be linked to the activation of autophagy in HCC. However, despite the autophagy induction, p62 expression was raised in sorafenib-resistant cells, which was associated with the enhanced oxidative stress and the upregulation of the transcription factor NRF2. It has been reported that p62 upregulation could correlate with lower sorafenib efficacy in HepG2 cells[43], being also related to dismal HCC prognosis[44]. Likewise, NRF2 overexpression has been related to poor prognosis in solid malignancies[45].These findings indicate that prolonged sorafenib treatment most likely activated the NRF2/p62 pathway, potentially conferring survival advantages.
Even though autophagy is frequently associated with chemoresistance, diverse HCC investigations accomplished with sorafenib described a death-promoting role of autophagy[41][46][47]. On the other hand, suppression of autophagy in vitro or in vivo has been shown to overcome drug resistance to oxaliplatin[36], doxorubicin[48], or cisplatin[49]. In the current research, abolition of the overactivated autophagy increased sorafenib-resistant cell death. Thus, in this case, autophagy is playing a cytoprotective action in sorafenib-resistant HCC and permits sorafenib-mediated cell death evasion. These findings are also supported by independent studies conducted with hydroxychloroquine[50], chloroquine[51], and 3-methyladenine, an inhibitor of autophagy at early phases[52]. Moreover, Liang et al.[22] and Lin et al.[21], two previous articles related to the topic of the current investigation, also showed that autophagy inhibition using 3-methyladenine promotes sorafenib anti-tumor effects under hypoxia.
Regarding the controversial and novel implication of FOXO3 in the autophagy-related sorafenib resistance, these two previous studies[21][22] explicitly reported contrary findings. Specifically, Liang et al.[22] detected that hypoxia induces transcriptional activity of FOXO3, thereby promoting autophagy and decreasing sorafenib efficacy in HCC. Oppositely, Lin et al.[21] defended that FOXO3 is downregulated in sorafenib-resistant HCC under hypoxia, leading to autophagy activation and sorafenib resistance.
In order to adequately study and clarify the involvement of FOXO3 in the development of sorafenib resistance associated with autophagy, it has been firstly evaluated in the present research the expression of FOXO3 in HCC patient samples, as well as the relationship with prognosis and autophagy pathway. In agreement with Ahn et al.[53] and Song et al.[54], which notified the association of FOXO3 overexpression with worse HCC phenotypes, FOXO3 levels have been also found increased in HCC samples and were related to lower survival rates. Moreover, autophagic genes expression positively correlated with FOXO3 expression in HCC samples, suggesting that FOXO3 high expression is a negative hallmark in HCC and is linked to autophagy. In vitro, sorafenib-resistant HCC cells also showed FOXO3 upregulation. This finding was also observed in the researches by Zhou et al.[18] and Liu et al.[19], which demonstrated that FOXO3 upregulation mediates the loss of sensitivity to doxorubicin, epirubicin, or cisplatin in HCC; but also in one investigation carried out with oxaliplatin-resistant HCC cells[55]. Likewise, Liang et al.[22] reported that hypoxia promotes nuclear retention of FOXO3 in non-resistant HCC cells, being linked to the loss of sorafenib sensitivity.
Next, to assess the direct implication of FOXO3 in the induced autophagy of sorafenib-resistant HCC cells, this factor was silenced, finding that FOXO3 knockdown suppressed autophagy and led to chemoresistant cell death. Therefore, these results prove that FOXO3 upregulation during the acquisition of resistance to sorafenib in HCC causes the activation of a cytoprotective autophagy that accounts for chemoresistant cell survival. Similarly, Liang et al.[22] denoted that hypoxia triggers FOXO3 activation, leading to autophagy induction and chemoresistance. However, this previous work was performed with conventional sorafenib-treated but non-resistant HCC models, which actually do not resemble the drug resistance phenomenon[22]. Therefore, results should be considered with caution. In addition, miR-223 has also been shown to target FOXO3-induced autophagy and enhance doxorubicin sensitivity in HCC[18], while osteopontin drived epirubicin and cisplatin resistance by the upregulation of FOXO3-dependent autophagy[19]. Similar reports are found in neuronal[17] and cervical cancer cells[20]. Contrariwise, Lin et al.[21] suggested that FOXO3 overexpression could improve n6-methyladenosine-related sorafenib sensitivity in HCC by autophagy inhibition under hypoxia.
Regorafenib is one of the principal second-line drugs used in HCC patients after sorafenib failure[56]. The pre-clinical efficacy of this TKI in non-resistant HCC in vitro and in vivo models has been already tested[31][57]; finding that regorafenib could even counteract the loss of sorafenib sensitivity mediated by hepatocyte growth factor in sensitive HCC cells[58]. Nevertheless, the impact of regorafenib on FOXO3 and autophagy in sorafenib-resistant HCC had not been evaluated yet. In this investigation, it has been demonstrated for the first time that regorafenib downregulates FOXO3 and impairs autophagy in sorafenib-resistant HCC. Moreover, it has been found that regorafenib also inhibits autophagy at last phases by blocking autophagosome-lysosome fusion. Collectively, these results proved for the first time that the anti-tumor actions of regorafenib as a second-line drug are also due to the abolishment of the FOXO3-mediated pro-survival autophagy, thereby supporting the implication of this cytoprotective mechanism in the development of sorafenib resistance.
In conclusion, this research disclosed that aberrant upregulation of FOXO3 and the consequent activation of a pro-survival autophagy plays a crucial role in the acquisition of resistance to sorafenib in advanced HCC. Therefore, FOXO3 is related to worse tumor phenotypes and dismal prognosis, representing a potential biomarker in HCC. Moreover, this investigation reported for the first time that regorafenib efficacy against sorafenib-resistant HCC also relies on the inhibition of such FOXO3 and autophagy upregulation. Hence, targeting FOXO3-mediated autophagy emerges as an interesting therapeutic approach in HCC to improve clinical efficacy of sorafenib.
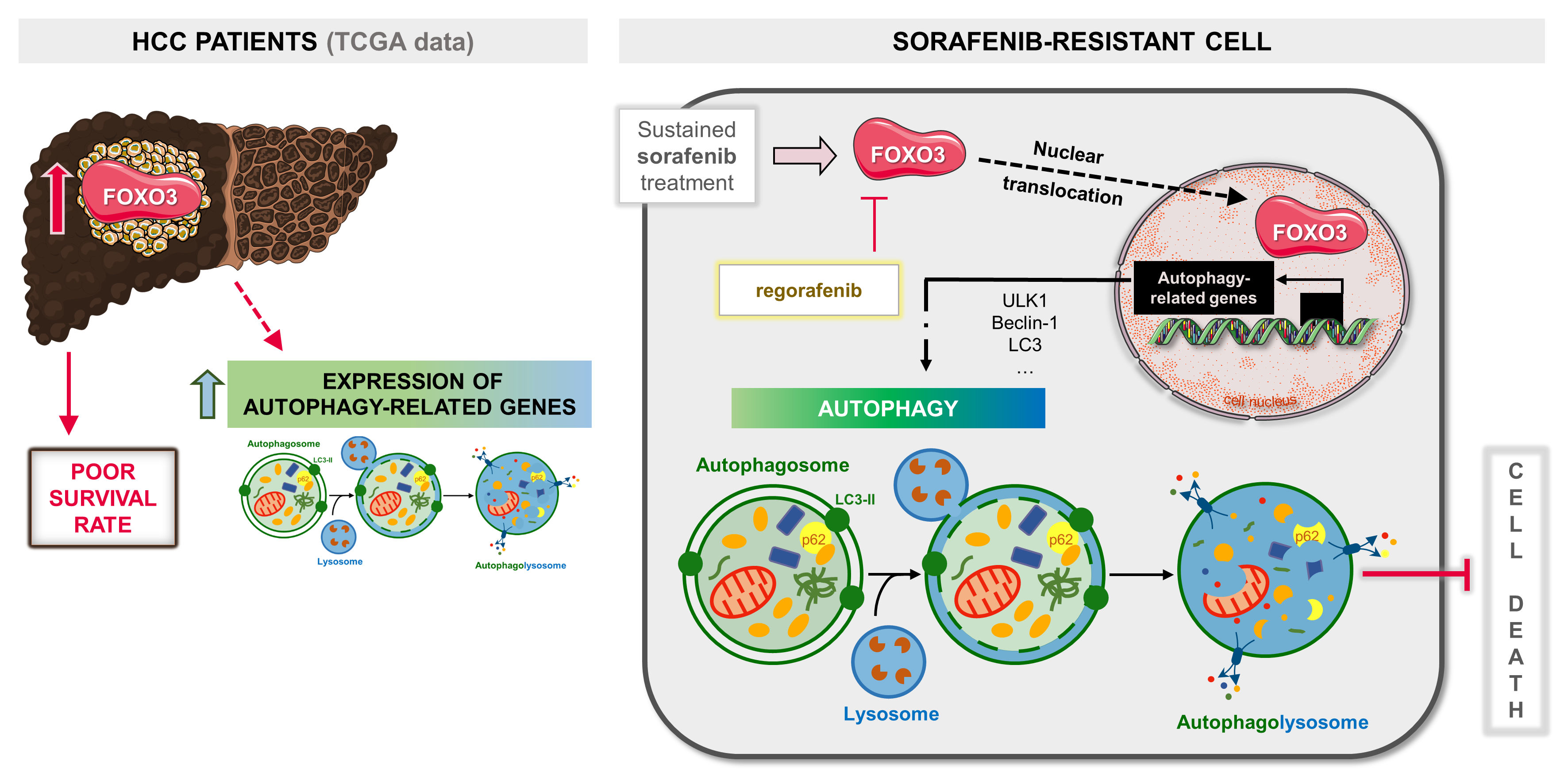
Graphical Abstract.
This entry is an adaptation from doi.org/10.3390/ijms222111770

