RNA interference (RNAi) uses small interfering RNAs (siRNAs) to mediate gene-silencing in cells and represents an emerging strategy for cancer therapy. Successful RNAi-mediated gene silencing requires overcoming multiple physiological barriers to achieve efficient delivery of siRNAs into cells in vivo, including into tumor and/or host cells in the tumor micro-environment (TME).
1. Introduction
The discovery of RNA interference (RNAi) in 1998 by Fire et al.
[1], laid the foundations for the development of new gene-targeting methodologies based on RNA oligonucleotides. More recently, the endogenous RNAi machinery in mammalian cells has been studied intensively, leading to the discovery of molecular mechanisms that allow for precise regulation of gene expression mediated by double-stranded RNA (dsRNA). DsRNAs, introduced into target cells using a delivery vector, are processed by Dicer, an RNAse III family member, which cleaves the dsRNA molecules into 19–23 nucleotide fragments that contain a 5′ phosphorylated end and an unphosphorylated 3′ end, with two unpaired nucleotide overhangs at each end. These small dsRNAs are called small interfering RNAs (siRNAs). The
N-domain unwinding activity of Argonaute (Ago)-2 unwinds the siRNA duplex into two single strands: the guide and passenger strands. Once unwound, the guide strand is incorporated into the RNA interference specificity complex (RISC), while the passenger strand is degraded. The RISC complex then binds to an endogenous mRNA that is complementary to the guide strand and cleaves the target mRNA through the separate endonuclease activity of Ago-2. These events affect the stability of target mRNAs leading to their degradation
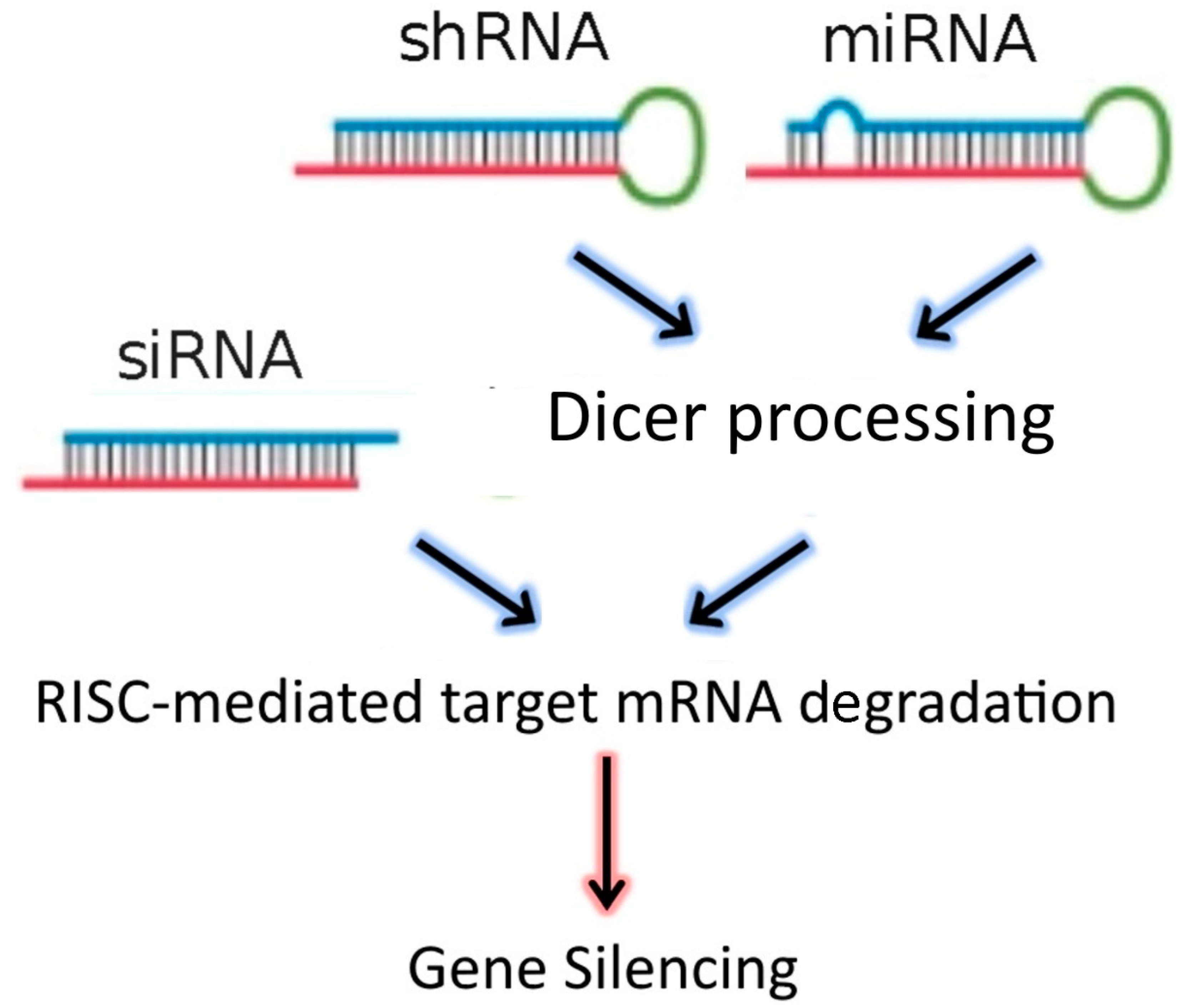
[2][3]. In addition to siRNAs, the dsRNA “targeting” sequences loaded into the RISC complex may also be derived from microRNAs (miRNAs), or from short-hairpin RNAs (shRNAs) (
Figure 1). MiRNAs are natural dsRNA molecules produced by all cells, which impact the function of many genes by blocking target mRNA translation
[4]. These RNA duplexes are produced from a stem-loop structure called the precursor miRNA and are processed into short dsRNAs by Dicer. Due to the short recognition length requirement, an individual miRNA is able to bind to multiple mRNAs, and hence it has the ability to regulate multiple genes due to reduced binding specificity. This also results in decreased efficiency of gene-silencing for any given gene, as compared to siRNAs. On the other hand, shRNAs are engineered in the laboratory as plasmids. RNA molecules with a tight hairpin turn are expressed from the plasmid, which can be used to facilitate long-term silencing of target gene expression via RNAi
[5]. Expression of an shRNA in cells may therefore typically be accomplished by intra-cellular delivery (for example, by transfection) of a plasmid containing specific shRNA sequences, able to target mRNA strands after being processed by Dicer. ShRNA plasmids have the additional advantage of being DNA-based, and so are more resistant to degradation than dsRNAs. However, shRNAs require the use of an expression vector, and so additional transcriptional steps are needed prior to the generation of dsRNA.
Figure 1. RNAi-based therapeutics for gene silencing.
Small interfering RNA (SiRNA), short-hairpin RNA (shRNA), and microRNA (miRNA) exert their activity in the cytoplasm of target cells, where they are incorporated into the RISC complex. However, in contrast to siRNAs, shRNAs and miRNAs must be previously processed by Dicer. After binding to the complementary mRNA sequence, Ago-2 mediates cleavage, and subsequent mRNA degradation. SiRNAs are exogenous dsRNAs, while miRNAs are derived from endogenous miRNA genes that are transcribed into primary miRNAs. ShRNAs are transcribed from a plasmid delivered to target cells.
By design, RNAi therapeutics can be targeted to facilitate the downregulated expression of specific genes, and RNAi is emerging as a form of treatment for a number of human diseases, including cancer. Multiple critical characteristics of tumor cells can, for example, potentially be targeted by specific RNAi therapies, aimed at reducing tumor burden and chemoresistance
[6][7][8]. However, the clinical application of RNAi therapy remains limited. A major reason for this is that siRNA therapeutics must overcome physiological and cellular barriers, hindering access of siRNAs to the cytoplasm of target cells (
Figure 2), where they are able to fulfill their regulatory function.
Figure 2. The intracellular barriers of siRNA-loaded NPs as nanovectors.
2. Challenges in siRNA Delivery: Physiological and Intracellular Barriers
In vivo delivery of siRNA has many challenges. Firstly, unmodified and unprotected siRNAs are unstable in serum, as they are easily degraded by RNAses
[9]. Multiple strategies that involve chemical modifications of the backbone or the bases of oligoribonucleotides have been used to protect siRNAs without impairing their capacity to bind target mRNA
[10]. Secondly, siRNAs injected into the bloodstream are very susceptible to removal by renal clearance, which results in a short siRNA half-life in blood
[11]. NP-based delivery systems have the ability to protect siRNAs from intravascular degradation and reduce the risk of degradation and/or interaction with non-target molecules. However, NPs need to be designed in ways to avoid a number of physiological barriers (
Table 1), which limits their ability to be delivered to target cells. For some delivery systems, the NP-based siRNA delivery systems are not required to reach the TME to be effective anti-cancer treatments. For example, cancer vaccines, which only need to be recognized by patrolling immune cells, can be injected subcutaneously. Lastly, irrespective of the target cell, siRNAs must be delivered to the cytoplasm of cells to fulfill their regulatory function and degrade target mRNA molecules, which necessitates the bypassing of the endosomal-lysosomal pathway.
Table 1. Physiological barriers in siRNA delivery by intravenous injection.
|
Barrier
|
Approach
|
|
Degradation by RNAses
|
Chemical modification of siRNAs, inclusion of siRNAs in NP-based delivery systems
|
|
Renal clearance
|
Inclusion of the siRNA in a nanocomplex with a HD >6 nm
|
|
Reticuloendothelial system
|
Addition of PEG to the nanocomplex to reduce protein corona formation and phagocytosis
|
|
Limited access into tumor tissue
|
Passive accumulation: limit NP size (<200 nm) to promote the EPR effect. Active targeting: Inclusion of a targeting ligand on the surface of the NPs
|
3. Lipid and Polymer-Based siRNA Carriers for Cancer Therapy
3.1. Liposomes
Liposomes are spherical vesicles composed of at least one lipid bilayer with an aqueous core. The liposomal membrane can be positively or negatively charged, depending on the phospholipid composition. SiRNAs can be incorporated into positively charged liposomes by electrostatic interactions forming lipoplexes
[12]. Cationic lipids including 1,2-dioleoyl-3-trimethylammonium propane (DOTAP),
N-[1-(2,3-dioleoyloxy) Propyl]-
N,
N,
N-trimethylammonium chloride (DOTMA) have been used in combination with neutral lipids such as cholesterol (Chol), DOPE, 1,2-dioleyl-sn-glycero-3-phosphocholine (DOPC), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DSPE) to form lipoplexes. In most cases, the siRNA–cationic lipid complexes have a much-reduced positive charge. However, electronegative or neutral liposomes have superior pharmacokinetic properties and are more bio-compatible than cationic ones. The inclusion of PEG chains dramatically reduces the positive charge on the surface of liposomes. On the other hand, PEG chains limit the uptake of lipoplexes by tumor cells, ultimately lowering their transfection efficiency
[13]. Additionally, PEG interferes with endosomal escape, resulting in siRNA degradation. The incorporation of a pH-sensitive molecular bridge between PEG and other components of the liposome can facilitate the endosomal release of siRNAs, increasing silencing efficiency. Shuian-Yin and colleagues have developed polymer-based liposomal complexes (SPLexes) composed of a pH-sensitive folate-PEG, carrying a vascular endothelial growth factor (VEGF)-targeting siRNA. The folate receptor CD44 is highly expressed in a wide variety of tumor types and can be exploited for specific tumor targeting. SPLexes showed an increased uptake by cancer cells expressing the folate receptor and were able to induce 75% downregulation of the target protein, demonstrating excellent translational potential
[14]. More recently, PEGylated DC-Chol/DOPE cationic liposomes were successfully used to treat ovarian cancer in a xenograft murine model. This formulation was designed to downregulate kinesin spindle protein (KSP) in ovarian cancer cells (HeyA8-MDR) to reduce paclitaxel (PDX) resistance. HeyA8-MDR tumor-bearing mice treated with liposomes, containing both PDX and the KSP-targeting siRNA (siKSP), showed reduced tumor growth compared to controls. In addition, KSP protein levels were highly downregulated in excised tumors, demonstrating the in vivo activity of siKSP. Biodistribution studies showed high levels of accumulation of liposomes in tumors and long half-life in the blood (16.5 h)
[15]. Another similar formulation was developed by Sheng Yu and colleagues, showing synergistic activity of PDX and Polo-like kinase 1 (PLK-1)-targeting siRNA in limiting the progression of breast cancer. These cationic liposomes were also functionalized with a targeting aptamer (AS1411) to further enhance tumor accumulation
[16]. In another report, the use of gemcitabine (Gem) in combination with Myeloid cell leukemia 1 (MCL1)-targeting siRNA in loaded liposomes was shown to be effective in treating pancreatic cancer
[17]. These experimental examples suggest that the use of siRNAs targeting specific oncogenes over-expressed in cancer cells are able to synergize with chemotherapy, resulting in reduced tumor growth in a wide variety of xenograft tumor models.
Specific antigen-targeting monoclonal antibodies (mAbs) can be coupled to liposomes in order to achieve specific cell targeting. However, the attachment of mAbs by covalent methodologies to the components of the lipidic bilayer is inefficient and requires careful optimization
[18][19]. Recently Kedmi and colleagues developed a flexible coupling technique called ASSET (anchored secondary scFv enabling targeting)
[20]. ASSET is a membrane-anchored lipoprotein that can be incorporated into lipoplexes, and by interacting with the antibody crystallizable fragment (Fc) domain of immunoglobulins, it enables the functionalization of an NP with potentially any antibody. With this methodology, the antibody variable domain is exposed for ligand binding, in contrast to standard coupling procedures, which can limit the functionality of the attached mABs. One of the formulations proposed was able to improve survival in a mantle cell lymphoma xenograft model. Another report by Guan et al.
[7], showed that active tumor targeting was not always necessary to achieve good therapeutic effects. The authors developed liposomes with a cationic core for siRNA loading and an outer layer composed of DSPE-PEG2000 to prolong circulation. These NPs, loaded with PDX and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-targeting siRNA, were devised to reduce chemoresistance and limit ATP production in tumors. Most tumor cells are characterized by enhanced glycolysis and high levels of GAPDH, which catalyzes the sixth catabolic reaction of glycolysis (conversion of glyceraldehyde 3-phosphate to D-glycerate 1,3-bisphosphate). Intravenous injections of siGAPDH-PDX liposomes led to a reduction in tumor burden in a murine xenograft model (Hela) with good accumulation in the TME, mediated by the EPR effect. Although this effect plays a critical role for NP accumulation in murine xenograft models, experimental evidence for the effectiveness of EPR in human tumors remains contradictory
[12].
3.2. PEI-Derived Nanosystems
Cationic polymers are attractive biomaterials for the complexation of nucleic acids, such as siRNAs. Polyethyleneimine (PEI) is one of the most studied, consisting of repeating units of the amine group and two carbon aliphatic CH
2CH
2 spacers, which is responsible for high levels of aqueous solubility and also for pH buffering capacity in the endosomal/lysosomal pathway. However, despite allowing efficient siRNA complexation through electrostatic interactions, PEI also exerts toxic effects on cells, depending on its structure and on the cell type tested
[21]. To reduce its toxicity, PEI has been conjugated to other polymers such as chitosan, hyaluronic acid (HA), cyclodextrins (CD), and PEG in order to form NPs that are able to protect siRNAs and facilitate endosomal escape.
Chitosan is a biodegradable, biocompatible, and non-toxic polymer, which has a low transfection efficiency for siRNAs
[22]. However, this drawback can be mitigated by coupling chitosan to PEI. For example, glycol-chitosan (GC)-PEI-siRNA NPs developed by Huh and colleagues exhibited strong tumor accumulation and target downregulation in vivo
[23]. Despite being a promising study, the authors did not go on to provide evidence of in vivo anti-tumoral activity, since the NPs were designed to carry an siRNA targeting red fluorescent protein (RFP), expressed by the xenograft tumors. Zhang et al., developed a dual-targeting chitosan-PEI nanosystem, incorporating the antineoplastic drug Lonidamine. The attached siRNA, targeting the apoptosis inhibitor protein Bcl-2, was covered by a layer of PEG-Poly(acrylic acid)-folic acid to prolong circulation and enhance cancer cell targeting. The mitochondria-targeting ligand triphenylphosphine (TPP) was incorporated into the chitosan-PEI polymer, enhancing the mitochondrial activity of Lonidamine. Although this formulation showed promising results in vitro, the analysis of its in vivo activity remains to be determined
[24].
HA is a polysaccharide composed of disaccharide units of
d-glucuronic acid, alternating with
N-acetyl
d-glucosamine (NAG), and it is a component of the ECM and synovial fluids. It is anionic, biodegradable, non-toxic, non-immunogenic, and has been used in targeting CD44, which is highly expressed by a variety of tumors
[25]. HA-PEI/PEG NPs have been developed by Ganesh and colleagues to carry siRNAs targeting the protein SSB and PLK1, demonstrating in vivo siRNA activity in xenograft tumor models. However, knockdown efficiency was highly dependent on tumor vascularization, suggesting that tumor accumulation was not solely mediated by the active targeting ligand HA
[26].
CD are cyclic oligosaccharides, consisting of a macrocyclic ring of glucose subunits joined by α-1,4 glycosidic bonds. β-CD nanosystems are characterized by a hydrophobic interior and hydrophilic exterior and they are used to enhance pharmacokinetics properties of loaded hydrophobic drugs whereas the cationic polyamine backbone allows an electrostatic interaction with siRNAs making them ideal candidates for synergistic drug delivery
[27]. Wang and colleagues developed a CD-PEI conjugate adsorbed to gold nanorods for the codelivery of docetaxel (DTX) together with siRNAs targeting the protein p65. The authors showed that the NF-κB pathway and its downstream cascade were inhibited by p65 blockade leading to an enhanced DTX effect and reduced tumor growth in vivo. Interestingly, the nanoplatform could facilitate tunable hyperthermia upon irradiation with a near-infrared (NIR) laser, thus triggering DTX release from the DTX-CD-PEI-siRNA complexes and inducing siRNA endosomal escape. Furthermore, the p65 knockdown sensitized 4T1 breast cells to DTX treatment by suppressing the expression of the anti-apoptotic gene, Bcl-2. The treatment of 4T1 tumor-bearing mice with DTX-CD-PEI-siRNA nanorods inhibited primary tumor growth and reduced the formation of lung metastasis
[28]. These results strongly suggest a synergistic effect of NIR irradiation, DTX, and p65 siRNA on tumor cells and the tumor vasculature, demonstrating considerable translational potential.
3.3. PLL-Derived Nanosystems
Similar to PEI, poly-L-lysine (PLL) is widely studied for the development of nanocarriers for nucleic acid delivery. PLL has a better biocompatibility and biodegradability profile than PEI
[29]. However, PLL/siRNA and PLL-PEG/siRNA polyplexes are more prone to interactions with serum proteins, reducing their ability to knock down target mRNAs
[30]. These findings suggest that serum albumin and other polyanions can compete with siRNAs for binding to PLL, leading to particle instability and siRNA disassembly. Interestingly, PLL derivatives are able to mitigate this drawback by enhancing serum stability. For example, polycaprolactone (PCL) has been used to develop a novel PEG-PCL-PLL polymer, which is able to bind siRNAs and form micelles
[31]. The assessed in vitro silencing efficiency for this formulation was similar to lipofectamine 2000 and superior to PEI. More recently, Xiao and colleagues developed PEG-PCL-PLL NPs, incorporating platinum-based chemotherapeutic agents (oxaliplatin and cisplatin) and Bcl-2-targeting siRNA for cancer treatment. In vitro experiments showed strong downregulation of Bcl-2 mRNA levels in MCF-7 and OVCAR-4 breast cancer cell lines. In addition, this formulation induced cell death was up to 100-fold more efficient than the free drugs in all the cancer cell lines tested
[32]. However, these NPs were not tested in murine tumor models.
PLL has also been conjugated to melanin, a biocompatible pigment, to take advantage of its excellent photothermal properties. Melanin generates heat under NIR irradiation
[33], which may be used to facilitate siRNA endosomal escape. The generated melanin-PLL polymer was loaded with
survivin-targeting siRNA and exhibited a strong inhibitory effect on 4T1 tumor cell growth, both in vitro and in vivo
[8]. Furthermore, the development of lung metastases was greatly reduced in treated mice compared to controls. In another report, Sun and colleagues developed a novel triblock polymer composed of poly aspartyl (
N-(
N′,
N′-diisopropylamino ethyl)) (PAD) conjugated to PLL-PEG. PAD was used to confer pH responsiveness to the polymer, thus enhancing the siRNA and DOX delivery to cancer cells. These NPs were loaded with DOX as an antineoplastic agent, and Bcl-2-targeting siRNA to induce apoptosis in cancer cells. Biodistribution analysis in HepG2/adriamycin (ADM) tumor-bearing mice showed that NP accumulation was observed at the tumor site 6 h after injection, and reached a maximum at 24 h, lasting for as long as 48 h. Furthermore, the treatment with DOX/siRNA-loaded PAD-PEG-PLL NPs was able to reduce tumor growth and increase the survival of tumor-bearing mice compared to controls. Lastly, ex-vivo analysis of the tumors showed reduced Bcl-2 and Ki67 expression after treatment, suggesting the induction of apoptosis and reduced cellular proliferation, which led to the control of tumor growth in the treated mice
[34]. Another similar nanosystem was recently developed by Wang and colleagues incorporating a disulfide bridge between PEG and PLL (PEG-SS-PLL) to induce PEG release in the endosomes and facilitation of siRNA delivery to the cytoplasm of cancer cells. A VEGF-targeting siRNA was included in the NP to reduce angiogenesis in the TME. In vivo efficacy of the formulation was demonstrated in a HepG2 xenograft murine model. PEG-SS-PLL-siVEGF NP treated mice showed reduced tumor growth compared to controls. Furthermore, histopathology and Western blot analysis of excised tumors showed reduced VEGF expression
[35].
3.4. Anti-Tumor Nanovaccines Enhanced by siRNAs
Therapeutic cancer vaccines aim to induce de-novo immune responses against cancer cells by promoting the activation and subsequent expansion of tumor-specific CD8
+ or CD4
+ T cells, which mediate anti-tumor immunity. NP-based cancer vaccines can help to improve antigen recognition and presentation by APCs (i.e., DCs). The incorporation of antigens in NPs can be achieved by covalent linkage of a protein or a peptide to components of the nanostructure. In addition, nucleic acids such as mRNA and DNA can be attached through electrostatic interactions to the surface of NPs (similarly to siRNAs) and can be processed and translated by APCs into antigenic peptides. Moreover, DNA and mRNA-based cancer vaccines are able to incorporate multiple antigens to increase immunogenicity and the activation of a strong and specific anti-cancer immune response. NP-mediated transfection with DNA or RNA coding for oncogenic proteins or peptides has the advantage of more closely mimicking live infections by incorporating multiple antigen epitopes into one construct. In addition, nucleic acids can serve as self-adjuvants, stimulating the endosomal toll-like receptors (TLR 3, 7, 8, or 9)
[36].
Not all NP-based cancer vaccines are required to reach the TME in order to be effective. Tumor targeting of nanovaccines can result in modification of the tumor immune infiltrate, due to the immunomodulatory activity of the adjuvant included in the NP, leading to enhanced anti-tumor responses
[37]. Nanovaccines are usually administered subcutaneously, where they form a depot, which causes local inflammation and infiltration of APCs able to take up and process the NP. After activation, DCs then migrate to the lymph nodes, where they present the antigen via the major histocompatibility complex (MHC) class I or II to CD8
+ or CD4
+ T cells, respectively. NPs can also be designed to drain directly into the lymphatic system without forming a local depot. For this purpose, NPs ranging from 30 to 100 nm have been shown to effectively reach lymph-nodes after subcutaneous injection while NPs of a larger size are unable to drain effectively into the lymphatic system and are retained at the injection site
[38].
The activity of cancer vaccines can be enhanced by the inclusion of siRNAs targeting one or more immune-related proteins aimed at further enhancing function and antigen presentation by APCs. Other siRNA targets include checkpoint blockade inhibitors, which dampen ongoing immune responses in the TME
[39]. Thus, including siRNAs in nanovaccines can further enhance the specificity of anti-tumor immunity. Recently, Huang and colleagues designed tumor-targeted lipid dendrimers for hepatocellular carcinoma (HCC) treatment. These NPs consisted of the antigenic molecule hemagglutinin (expressed by the implanted HCC cell line in mice), a PD-L1 siRNA, and an IL-2 expressing plasmid to enhance effector T cell activity. These NPs provide adjuvant activity by inducing the STING pathway, which triggers the secretion of inflammatory cytokines such as CCL5, CXCL10, and IFN-β to further enhance immune cell activity in the TME. In vivo experiments on HCC murine xenografts demonstrated increased tumoral infiltration of CD8
+ T cells, primary tumor growth suppression, and inhibition of distal metastasis
[40].
A microparticle formulation derived from the bacteria, Propionibacterium acnes, called MIS416, was covalently attached to the model antigenic peptide, SIINFEKL, to study its potential as a cancer vaccine. The adjuvant properties of MIS416 were conferred through its cell wall skeleton, consisting of immunostimulatory muramyl dipeptide repeats and CpG sequences which, respectively, activated NOD-2 and TLR-9 receptors to induce DC activation. SIINFEKL was conjugated to MIS416, utilizing a streptavidin bridge between biotinylated versions of MIS416 and SIINFEKL. This formulation was able to enhance costimulatory molecules on treated DCs and induced strong antigen presentation on MHC molecules. Furthermore, in vivo cytotoxicity experiments with the MIS416-SIINFEKL conjugate resulted in the induction of a specific anti-SIINFEKL immune response
[41]. In a follow-on study, the feasibility of using MIS416 to deliver signal transducer and activator of transcription
3 (STAT3)-targeting siRNAs was further explored to enhance DC function. A biotinylated STAT3 siRNA, which was conjugated to MIS416 through a disulfide linkage, allowed endosomal escape of the siRNA, and following the treatment of DCs with MIS416-SS-siStat3 the downregulation of both STAT3 mRNA and STAT3 protein levels were observed compared to controls. These studies suggest that an siRNA gene targeting approach could potentially be used to further enhance the cancer vaccine capabilities of MIS416
[42].
 +1 credit
+1 credit