Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicolas DUBUISSON | + 4011 word(s) | 4011 | 2021-11-09 09:18:30 | | | |
| 2 | Lindsay Dong | Meta information modification | 4011 | 2021-11-29 09:51:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dubuisson, N. NLRP3 Inflammasome and Skeletal Muscle. Encyclopedia. Available online: https://encyclopedia.pub/entry/16379 (accessed on 07 February 2026).
Dubuisson N. NLRP3 Inflammasome and Skeletal Muscle. Encyclopedia. Available at: https://encyclopedia.pub/entry/16379. Accessed February 07, 2026.
Dubuisson, Nicolas. "NLRP3 Inflammasome and Skeletal Muscle" Encyclopedia, https://encyclopedia.pub/entry/16379 (accessed February 07, 2026).
Dubuisson, N. (2021, November 25). NLRP3 Inflammasome and Skeletal Muscle. In Encyclopedia. https://encyclopedia.pub/entry/16379
Dubuisson, Nicolas. "NLRP3 Inflammasome and Skeletal Muscle." Encyclopedia. Web. 25 November, 2021.
Copy Citation
Nucleotide-binding oligomerization domain (NOD-), leucine-rich repeat (LRR-) and pyrin domain-containing protein 3 (NLRP3) is a protein coded by the Nlrp3 (CIAS1) gene and is composed of an amino-terminal pyrin domain (PYD), a central nucleotide-binding domain (NACHT), and a C-terminal leucine rich repeat (LRR) motif. To date, NLRP3 has been extensively studied in the heart, where its effects and actions have been broadly documented in numerous cardiovascular diseases.
skeletal muscle
NLRP3
inflammasome
1. Introduction
Recently, increasing evidence has highlighted skeletal muscle as having an active and pivotal role in the immune response [1][2][3]. The main evidence leading to this assertion was the ability of muscle cells to secrete their own cytokines, known as myokines. These myokines consist of several hundred secreted proteins and peptides, which may act locally or systemically to mediate numerous metabolic and immune responses [4][5]. Moreover, the detection of the mRNA expression levels of all innate immune receptors in human skeletal muscle biopsies, isolated muscle fibers and primary myotubes confirmed their active involvement as immune effectors [6]. We, too, have provided first evidence for the presence of formed and active NLRP3 within skeletal muscle fibers. NLRP3 inflammasomes were indeed detected as stained clusters in the sarcoplasm of myofibers from wild type (WT) mice challenged by LPS, but not from Nlrp3 knockout mice [3].
In normal conditions, once activated, the NLRP3/caspase-1/IL-1β pathway activation is a defense mechanism leading to an antiviral, antibacterial, antifungal and anti-inflammatory response that is programmed to cure the pathological tissue [7][8][9][10]. Indeed, in virus mediated disease models, Nlrp3-KO mice developed more severe disease than infected WT animals [9]. NLRP3 activation has also been shown to induce leukocyte aggregation and efficient inflammatory responses in Aspergillus fumigatus infected mice [10].
On the other hand, excessive NLRP3 inflammasome activation has been tightly associated with several disorders involving skeletal muscle alterations.
2. NLRP3 and Skeletal Muscle Diseases
This section is dedicated to muscle diseases where NLRP3 has been described to play a key pathogenic role (Figure 1).
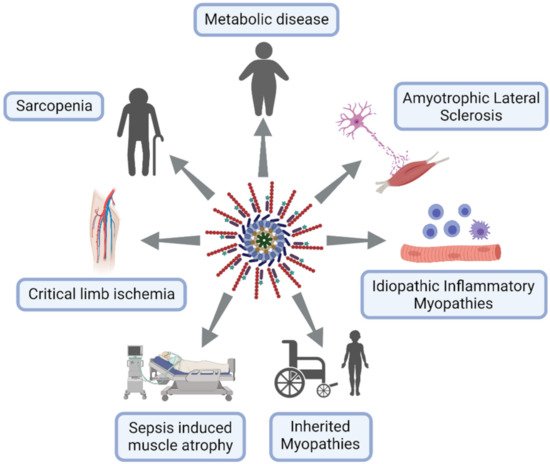
Figure 1. NLRP3 inflammasome excessive activation in skeletal muscle diseases. Lymphocytes in blue, macrophages in purple and neuron in pink.
2.1. Metabolic Disorders
Metabolic syndrome (MS) encompasses several disorders involving obesity, insulin resistance and type 2 diabetes, hypertension and dyslipidemia leading to cardiovascular disease. Other associated burdens result from ectopic lipid deposits giving rise in liver to non-alcoholic fatty liver disease progressing into non-alcoholic steatohepatitis (NAFLD/NASH), and, in skeletal muscle, to myosteatosis. Hyperuricemia is also often present in this syndrome [11]. All of these components are characterized by chronic low grade inflammation potentially mediated by inflammasomes [12][13][14][15]. The NLRP3 inflammasome may, therefore, be a link between metabolism and inflammation, strengthening the recent concept of metainflammation.
NLRP3 may be a sensor for metabolic “danger” signals, which may either serve as priming signals that induce NLRP3 and pro-IL-1β transcription or as second phase signals triggering inflammasome formation [14][15] (Figure 2).
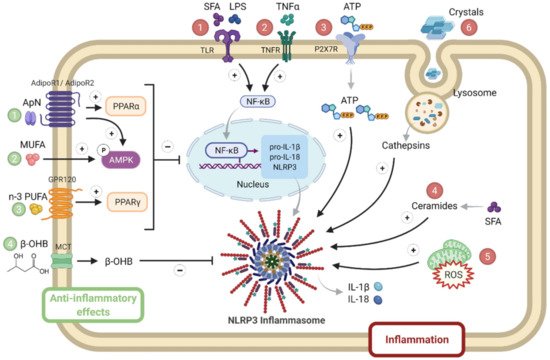
Figure 2. NLRP3 priming and activation in metabolic syndrome. Several activators of either priming or NLRP3 complex formation are numbered in red. They include SFA, LPS ①, inflammatory cytokines such as TNFα ②, stress molecules such as ATP ③, ceramides ④ and ROS ⑤, as well as crystals ⑥. By contrast, inhibitory hormonal or metabolic signals are numbered in green: they include adiponectin (1), MUFA (2) or n-3 PUFA (3), or ketone bodies (β-OHB) (4). Adiponectin binds to AdipoR1 (predominantly expressed in skeletal muscle) or to AdipoR2 (predominantly expressed in liver) to activate AMPK or PPAR-α signaling, respectively. MUFA and PUFA activate AMPK and PPAR-γ signaling, respectively. β-OHB is produced by the liver and is used as fuel by other tissues (such as muscle), where it enters via a MCT. β-OHB, β-hydroxybutyrate; MCT, monocarboxylate transporter; MUFA, mono unsaturated fatty acids; PPAR, peroxisome proliferator activated receptor; PUFA, poly unsaturated fat; SFA, saturated fatty acids. Symbols in circles indicate: ‘P’, phosphorylation; ‘+’, activation; and ‘−’, inhibition.
Priming signals act through the innate immune receptor (toll-like receptor 4), whose ligands include LPS and saturated fatty acids (SFA) [16], or through inflammatory cytokine receptors. Levels of SFA are often increased in MS due to the alleviation of the antilipolytic action of insulin [17], as well as circulating LPS due to dysregulated microbial colonization and ensuing increased gut permeability with the translocation of endotoxins [18]. Priming signals lead to enhanced NF-κB signaling. Like priming signals, second phase signals are also elevated in MS: these include stress molecules such as ATP, ROS and ceramides that activate NLRP3 formation [12][13][14]. Ceramides constitute a subtype of sphingolipids, which are increased in plasma, adipose tissue, liver and the skeletal muscle of animal models with insulin resistance and patients with MS. Ceramide levels negatively correlate with insulin sensitivity [19]; some ceramides may derive from SFAs [14]. Eventually, uric acid can form crystals that are well known NLRP3 activators [13]. By contrast, some molecules may physiologically taper inflammasome responses: adiponectin [5], mono or poly unsaturated fat (n3) [14] or ketone bodies (β-hydroxybutyrate, β-OHB) [20] (Figure 2).
The adipocyte hormone, adiponectin, could simultaneously thwart several facets of the metabolic syndrome by its insulin-sensitizing, fat-burning and anti-inflammatory/antioxidative properties. Adiponectin binds to its receptors: AdipoR1, mainly expressed in skeletal muscle, or AdipoR2, mainly expressed in liver, to activate AMPK or peroxisome proliferator activated receptor (PPAR)-α signaling, respectively, thereby inducing its biological responses. However, adiponectin levels are decreased in obesity and in patients meeting the criteria for the metabolic syndrome [5]. A balanced diet should contain mono or poly unsaturated fat (n3), which may inhibit inflammasome through AMPK or PPAR-γ. Unlike caloric excess, energy deficit, such as starvation, generates metabolic signals, such as β-hydroxybutyrate, that may dampen innate the immune response, which results in sparing energy for the major ketone dependent organs, such as the brain and heart [20] (Figure 2).
Most of the first and second phase signals that enhance NLRP3 responses may either activate the transcription of inflammatory genes via NF-κB or inhibit key components of the insulin-signaling cascade through inactivating phosphorylation, thereby promoting insulin resistance [12][21]. In obesity, adipocyte hypertrophy induces cellular stress together with immune cell infiltration, the release of inflammatory adipokines and insulin resistance. Once adipose tissue storage capacity is overwhelmed, ectopic lipid deposit occurs in several tissues, including the liver and skeletal muscle, further worsening inflammation and insulin resistance [17]. This leads to NAFLD/NASH in the liver and to myosteatosis in skeletal muscle. NLRP3 plays a crucial role in metainflammation and in the interplay between these three insulin target tissues (Figure 3).
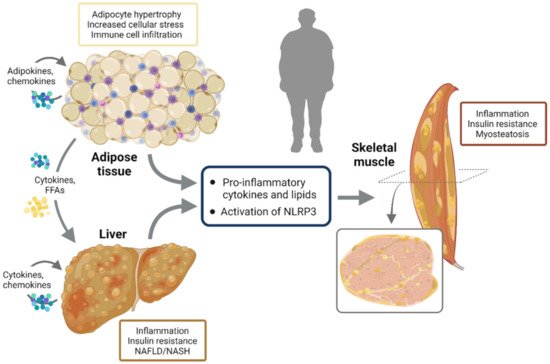
Figure 3. NLRP3 activation and interplay between insulin target tissues. In obesity, adipocyte hypertrophy induces cellular stress together with immune cell infiltration, release of inflammatory adipokines and insulin resistance. Once adipose tissue storage capacity is overwhelmed, ectopic lipid deposit occurs in several other tissues including the liver and skeletal muscle, further worsening inflammation and insulin resistance. This leads to NAFLD/NASH in the liver and to myosteatosis in skeletal muscle. NLRP3 plays a crucial role in metainflammation and in the interplay between these three insulin target tissues.
2.2. Muscle in Aging/Sarcopenia
As society ages, the incidence of physical limitations is dramatically increasing, thereby enhancing the risk of falls, institutionalization, comorbidity, and premature death. An important cause of physical limitations is the age related loss of skeletal muscle mass and function, also referred to as sarcopenia [22][23]. Beyond physical performance, muscles also play a crucial role in insulin sensitivity and fuel homeostasis. Muscle disturbances may, thus, lead to insulin resistance and metabolic disorders [24].
Overall loss of skeletal mass results from an imbalance between muscle protein anabolic and catabolic pathways, where protein synthesis is hindered and protein breakdown is excessive [25]. However, the cellular and molecular mechanisms underlying sarcopenia are still poorly understood, although this condition is currently considered to be multifactorial [26]. The aging process is associated with a decline in autophagic capacity which impairs cellular housekeeping, leading to protein aggregation and the accumulation of dysfunctional mitochondria, which provoke ROS production and oxidative stress. These danger signals, in turn, activate inflammasomes which provoke a low grade inflammation in several tissues, referred to as inflammaging. This further inhibits autophagy and accelerates the aging process [27][28]. Chronic inflammation together with reduced muscle mass could also promote insulin resistance. Insulin resistance, inflammatory cytokines, the inhibition of autophagic capacity, mitochondrial dysfunction and ROS induce and perpetuate inflammaging and lead to sarcopenia [29]. In addition, inflammaging could aggravate several other age related degenerative changes [27][30].
The involvement of inflammasomes in ageing and age related diseases [31], including sarcopenia, has been strengthened by studies in mice with genetic deletion of Nlrp3. Thus, deletion of the NLRP3 inflammasome enhances healthspan and protects against insulin resistance, bone loss, reduced cognitive function and motor performance [32]. Ageing is also associated with decreased skeletal muscle strength and slowing of movement, in which increased NLRP3-dependent caspase-1 activity in muscle is described. The deletion of mouse Nlrp3 prevented the reduction in muscle mass, increased muscle strength and endurance and protected from age related increases in the number of myopathic fibers [33]. Another study confirmed these data and further showed a reduction in fibrosis and apoptotic nuclei in the skeletal muscles of aged Nlrp3-KO mice, compared to wild-type ones, as well as less mitochondrial damage and multivesicular bodies resulting from defective autophagy/mitophagy [34]. Oral administration of melatonin, a pineal hormone with antioxidant and anti-inflammatory properties, in a mouse model of sarcopenia was also shown to reduce the expression of pro-caspase 1 mRNA and to preserve the normal muscular structure and activity of skeletal muscles [35][36][37].
2.3. Critical Limb Ischemia
Critical limb ischemia (CLI) is the most severe clinical manifestation of peripheral arterial disease, where failure to establish revascularization eventually leads to amputation and even to death [38]. Metabolic syndrome and ageing, both characterized by low grade inflammation, are risk factors for the development of peripheral arterial disease and are known to impair skeletal muscle postischemic vascular recovery [39]. However, the molecular mechanisms involved are still unknown. A hypothesis suggests that NLRP3 could also be implicated in mediating inflammation and angiogenesis after ischemia. In a hind limb ischemia mouse model, blood flow and vascular density were impaired in HFD mice. This was carried out through TXNIP-dependent NLRP3 inflammasome activation in muscle, which led to significant increases in active caspase-1 and IL-1β and compromised vascular recovery in response to ischemia. Targeting the NLRP3 inflammasome by using Txinpt-KO mice mitigated HFD-induced inflammation and impaired angiogenesis, thus opening a potential therapeutic target in obesity induced vascular complications [40].
Ischemic murine muscle also exhibited a reduced expression of a specific circular RNA (circHIPK3). Treatment with exosomes delivering circHIPK3 into skeletal muscle reduced ischemia induced pyroptosis caused by inflammasome, as evidenced by less activation of NLRP3, cleaved caspase-1, and reduced increase in IL-1β and IL-18.
Finally, heme oxygenase-1 (HO-1) could also be a critical player in inducing NLRP3 in ischemic muscle. While postischemic inflammation is needed for initiation of neovascularization, excessive inflammatory response suppresses perfusion recovery. HO-1 is an immunomodulatory enzyme primarily expressed in macrophages [41].
2.4. Sepsis Induced Muscle Atrophy
Sepsis is an excessive response of the body against an infection leading to tissue and organ damage. This condition requires intensive care management and accelerates muscle atrophy in bed bound patients [42]. Muscle wasting is linked to the inflammatory response occurring during the acute phase of sepsis, and might potentially be mediated by the NLRP3 inflammasome [43].
The skeletal muscles of mice subjected to acute inflammation by intraperitoneal LPS injection displayed significantly high levels of inflammatory components, such as the NLRP3 inflammasome and IL-1β. This was accompanied by an increase in muscle atrophy signaling pathways. The Forkhead box O (FoxO) family of transcription factors plays a critical role in protein breakdown by activating the expression of atrogenes (which include two muscle specific ubiquitin ligases, atrogin-1 and MuRF1) responsible for profound loss of muscle mass. All of these components were upregulated in the muscles of LPS mice. These deleterious effects were abolished in mice pretreated with an inhibitor of a double strand RNA-dependent protein kinase (PKR), which blocks inflammatory cytokine expression [44]. Likewise, Triptolide, a plant derivative previously described as an NLRP3 inhibitor [45], attenuated LPS-induced myotube atrophy in vitro in C2C12 cells and in vivo in LPS injected mice. Thus, triptolide decreased plasma inflammation while increasing skeletal muscle weight, strength and locomotion, thereby preventing muscle atrophy in LPS challenged mice [46]. OLT1177, an orally active β-sulfonyl nitrile molecule targeting the NLRP3 NACHT domain, was also shown to reduce the severity of systemic inflammation in mice challenged with LPS, where muscle IL-1β and oxidative stress were lowered [47]. Moreover, Nlrp3-KO mice, submitted to polymicrobial sepsis induced by cecal ligation and puncture surgery, had a survival benefit and did not lose body or muscle weight during 96 h of sepsis, when compared to wild type ones. This was associated with a reduction in IL-1β serum levels [48].
Finally, blocking gasdermin D pore formation by Disulfiram treatment (an approved drug used to treat alcohol addiction, see last chapter) tapered LPS induced sepsis in mice: circulating levels of inflammatory cytokines were reduced and survival was greatly improved. One advantage of this drug is to block LPS induced inflammasome activation by both noncanonical and canonical pathways [49].
2.5. Inherited Myopathies
Inherited myopathies are a heterogeneous group of diseases primarily affecting the skeletal muscle tissue. These are caused by mutations in different genes encoding proteins that are critical for muscle structure and function [50] (https://rarediseases.info.nih.gov/, accessed on 12 October 2021). They are characterized by progressive muscle weakness and wasting, along with a severe and persistent muscle inflammation that plays a central role in the onset and progression of these diseases [5]. Various studies have demonstrated that the NLRP3 inflammasome triggers a pathogenic inflammatory response in many inherited myopathies, including limb girdle muscular dystrophy type 2B (LGMD2B) [1], valosin-containing protein (VCP) associated diseases [2], and Duchene muscular dystrophy (DMD) [3][51].
LGMD2B is one type of limb-girdle muscular dystrophy, a group of heterogeneous diseases that affect the voluntary muscles. LGMD2B is caused by mutations in the dysferlin gene, which encodes a protein that is thought to aid in repairing the muscle fiber membrane when it becomes damaged or torn. LGMD2B is a slowly progressive disease that causes muscle weakness and atrophy, mainly of the pelvic muscles and muscles of the shoulder girdle [52] (https://rarediseases.info.nih.gov/, accessed on 12 October 2021).
VCP is a newly identified calcium associated ATPase protein that has been associated with various degenerative disorders that encompass inclusion body myopathy, Paget’s disease of bone, and frontotemporal dementia. VCP disease is a rare and progressive neuromuscular disorder, with death typically occurring in the 50s and 60s from respiratory and cardiac failure [53] (https://rarediseases.info.nih.gov/, accessed on 12 October 2021).
DMD is the most frequently inherited human myopathy and the most devastating type of muscular dystrophy. DMD is caused by mutations in the gene encoding for dystrophin, a key scaffolding protein, which forms an important protein complex that connects the actin cytoskeleton of myofibers to the extracellular matrix (Figure 3). This complex is crucial for maintaining cell membrane stability and permeability, as well as normal contractile function of the skeletal muscle. Absence of dystrophin leads to the disruption of this complex and, thus, to membrane damage, allowing for DAMP release, chronic inflammation and severe muscle degeneration. DMD remains a lethal muscle disorder with no cure, where the first signs of muscle weakness begin early on in life and, without proper intervention, death typically occurring in the 20s and 30s [5][54].
In addition, activating AMPK leads to an upregulation of the adiponectin muscle anti-inflammatory mediator, miR-711. We found that adiponectin, through miR-711, could be a major repressor of the NLRP3 inflammasome by inhibiting both its priming and activation in muscle [3][5]. This repression occurred both in vitro, in C2C12 myotubes, and in vivo after either local or systemic adiponectin supplementation [3][55] (Figure 4). In agreement with our data, other drugs and molecules that mainly activate AMPK signaling, such as AICAR (analog of adenosine monophosphate), resveratrol, a natural polyphenolic compound, and metformin (used for treating type 2 diabetes) were also found to mitigate some features of DMD in cell cultures and in animal models [56][57][58][59]. Metformin was also tested in a randomized, double-blind, placebo-controlled Phase III clinical trial in combination with L-citrulline, as a possible treatment for DMD. The study is completed and it showed only a small reduction in motor function decline among the stable subgroup of patients treated with this combination therapy (ClinicalTrials.gov Identifier: NCT01995032, accessed on 2 November 2021). A big part of their beneficial effects is mediated through the reduction in inflammation and in the inflammasome [31][54]. Similar to adiponectin, the gastric peptide ghrelin, famously known as the “hunger hormone”, is another circulating hormone with an anti-inflammatory effect [60]. Once injected in mdx mice, ghrelin was found to improve muscle performance and alleviate muscle pathology through the inhibition of NLRP3 inflammasome activation and subsequent maturation of IL-1β [51]. In addition, the plant compound, curcumin, a NF-κB inhibitor, also showed beneficial effects on the dystrophic skeletal muscle by reducing the levels of TNFα and Il-1β and improving cell membrane integrity, once injected in mdx mice [61] (Figure 4).
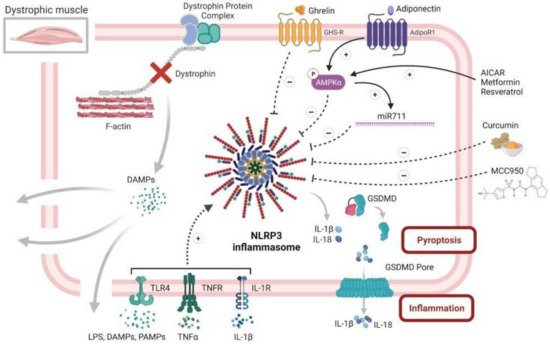
Figure 4. NLRP3 inflammasome and diseased skeletal muscle, case of Duchenne muscular dystrophy. This figure summarizes the central role of NLRP3 in dystrophic muscle inflammation, as well as several of its potential inhibitors. Briefly, the dystrophic muscle is characterized by absence of the dystrophin protein and its associated protein complex, which, in normal cells, links the intracellular cytoskeleton to the extracellular matrix, thus preserving the fiber membrane stability and permeability. Simple contraction of the dystrophic muscle causes microtears in the membrane and the subsequent release of intracellular DAMPS, which, in turn, can act in an autoparacrine manner to activate NLRP3 inflammasome in muscle fibers. Activated NLRP3 leads to an extreme release of inflammatory cytokines and DAMPS, thus maintaining and exaggerating the inflammatory response, eventually leading to pyroptosis and muscle degeneration. Rescuing the dystrophic phenotype can be achieved by alleviating muscle inflammation, in part through repression of NLRP3 inflammasome. This action can be accomplished by several factors. Firstly, adiponectin, a pleiotropic adipokine with potent anti-inflammatory effects, could strongly activate AMPK pathway that represses NLRP3 through reducing NF-κB activity and oxidative stress. Secondly, adiponectin, also through AMPK, increases the expression of its muscle anti-inflammatory mediator, miR-711, which can inhibit both the priming and activation of NLRP3. Thirdly, several drugs and molecules, such as AICAR, resveratrol and metformin, could repress NLRP3 through specific activation of the AMPK pathway. Fourthly, ghrelin, a gastric peptide with anti-inflammatory effect, could also put a brake on muscle inflammation through reduction in NLRP3 activation. Fifthly, curcumin, a potent NF-kB inhibitor, could hinder NLRP3 and the production of inflammatory cytokines. Finally, MCC950, a specific NLRP3 inhibitor, could greatly attenuate the pathogenesis of the dystrophic phenotype, mainly by protecting the muscle from inflammation. Symbols in circles indicate: ‘P’, phosphorylation; ‘+’, activation; and ‘−’, inhibition.
Finally, we are currently investigating a possible beneficial and therapeutic effect of MCC950, a specific NLRP3 inhibitor, on the pathogenesis of DMD using both in vivo mdx mice and in vitro primary cultures of human DMD myotubes. Preliminary results look promising and show an improvement in muscle performance and protection against muscle inflammation [62] (Figure 4).
2.6. Acquired Myopathies
Idiopathic inflammatory myopathy (IIM) is an acquired immune mediated muscle disease including dermatomyositis (DM), polymyositis (PM), sporadic inclusion body myositis (sIBM) and juvenile dermatomyositis (JDM) [63]. IIM is characterized by myalgia, muscle weakness, and extramuscular manifestations. Muscle biopsies show alterations such as necrosis, atrophy and sometimes inflammatory infiltrates [64][65][66].
To date, the pathogenesis of IIM remains unclear. However, several pieces of evidence indicate that the NLRP3 inflammasome may be involved in muscle damage. Recent studies have shown the direct impact of the canonical and noncanonical pathways of pyroptosis in the occurrence and progression of IIM [67][68]. In the experimental autoimmune myositis (EAM) mice model, serum levels of IL-1β and IL-18, as well as mRNA expression and the protein levels of NLRP3, GSDMD, Caspase 11 and P2X7R were increased [67]. The implication of the NLRP3 inflammasome has also been confirmed in humans, where IL-1β and IL-18 were shown to be highly expressed in the muscle and serum of DM and PM patients [69][70][71]. Moreover, DM and PM patients displayed a higher protein expression of NLRP3 and caspase-1 in muscle tissues, in comparison with controls [69][71]. Finally, N-GSDMD, the executioner of pyroptosis, was also upregulated in PM patients [66]. Taken together, these results confirm the potentially pivotal regulatory role of the NLRP3 inflammasome in IIM genesis and progression.
Several molecular pathways have been proposed to explain NLRP3 inflammasome activation in IIM pathogenesis. Firstly, TNFα activates NF-κB signaling leading to the canonical activation of the NLRP3 inflammasome [72]. Secondly, ROS trigger the activation of the NLRP3 inflammasome through TXNIP and the release of mitochondrial DNA [73]. Thirdly, mTOR pathway, via mTORC1 activation, stimulates IL-1β expression and maturation through hypoxia inducible factor-1α (HIF-1α), while rapamycin, a selective inhibitor of mTORC1, reduces NLRP3 and IL-1β levels [67]. Moreover, hypoxia upregulates the high mobility group box 1 protein (HMGB1), leading to the activation of NF-κB pathway and of NLRP3/IL-1β axis, thus triggering an inflammatory response [67].
Finally, glucose metabolism dysregulation could also contribute. Indeed, 18-fluorodeoxyglucose positron emission tomography/computerised tomography (PET/CT) showed abnormal glucose uptake in muscle tissues of patients with IIMs [74]. These observations were then explained by the direct implication of the glycolysis in the muscle damage process in IIM. Indeed, upregulation of pyruvate kinase isozyme M2 (PKM2) was shown in DM and PM compared with controls. Moreover, muscle PKM2 expressions were correlated with NLRP3 inflammasome expression levels [66], confirming a relationship between NLRP3 and dysregulated glucose metabolism [75].
Taken together, these results suggest that NLRP3 is involved in the immune response and myofiber alteration in IIMs.
2.7. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by progressive muscle weakness and atrophy, ultimately leading to death within approximately 2 to 5 years. The exact pathogenesis of ALS is still unknown. However, inflammation has been shown to be a prominent pathological finding in ALS patients [76] and a few studies suggest that the NLRP3 inflammasome might have a pivotal role in ALS. Indeed, the activation of the NLRP3 inflammasome has been observed, in brain, spinal cord and in the skeletal muscle of SOD1G93A mice, a transgenic mouse model of ALS expressing a mutant form of human Superoxide Dismutase 1 (SOD1), [77][78], as well as in those of sporadic ALS (sALS) patients [79][80][81]. NLRP3 mRNA levels were also significantly elevated in the white blood cells of sALS patients, compared to healthy controls [82]. In addition, mRNA levels of ASC, caspase 1 and IL-1β were increased at the asymptomatic stage in skeletal muscles of SOD1G93A mice [82][81], whereas their respective protein expression was still normal. However, in the later stage of the disease, increased protein levels of inflammasome components were observed [82]. Therefore, a link between NLRP3 inflammasome activation and ALS disease progression is thought to exist.
This early involvement of muscle revised our preconceived idea, in which muscle alterations are only the consequence of motoneuron destruction. Regarding the current data, skeletal muscle is increasingly considered as an active player in ALS pathogenesis. Indeed, as explained before, skeletal muscle expresses different PRRs, allowing a muscle specific response to environmental factors [6][83]. Moreover, primary muscle cells may release IL-1β after treatment with LPS and ATP confirming its primary role in inflammasome activation [1][3]. Muscle inflammation might then activate a retrograde signaling, leading to motoneuron death [84][85][86].
Interestingly, the NLRP3 inflammasome may play a dual role in ALS pathogenesis. Indeed, at an early stage of the disease, the NLRP3 inflammasome may exert positive effects. Thus, a positive correlation was observed between Nlrp3 mRNA levels in skeletal muscle and lifespan in SOD1G93A mice [82]. This might be partially explained by the fact that NLRP3, independently from its role in inflammasome, can act as a transcription factor in Th2 lymphocytes, promoting the expression of IL-4 [87], a cytokine responsible for muscle growth and regeneration [88]. The positive effect of NLRP3 on mouse lifespan was confirmed, as mice that did not receive MCC950, a selective inhibitor of NLRP3, lived longer [82]. These paradoxical results display the early role of the NLRP3 inflammasome, which is to clear noxious protein aggregates, a characteristic feature of ALS [89].
Taken together, these results suggest the two faced action of the NLRP3 inflammasome in ALS: where, at the early stage of the disease, it plays a beneficial role by clearing noxious aggregates, while, when the disease progresses, the chronic NLRP3 stimulations by an excess of damage signals, such as mutant proteins SOD1 and TDP-43, change its positive effect into a harmful action leading to myofiber damage and, ultimately, to motoneuron degeneration [89][90].
References
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-Regulation and Activation in Dysferlin-Deficient Skeletal Muscle. Am. J. Pathol. 2010, 176, 2891–2900.
- Nalbandian, A.; Khan, A.A.; Srivastava, R.; Llewellyn, K.J.; Tan, B.; Shukr, N.; Fazli, Y.; Kimonis, V.E.; BenMohamed, L. Activation of the NLRP3 Inflammasome Is Associated with Valosin-Containing Protein Myopathy. Inflammation 2017, 40, 21–41.
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 Inflammasome by Adiponectin Rescues Duchenne Muscular Dystrophy. BMC Biol. 2018, 16, 33.
- Lecompte, S.; Abou-Samra, M.; Boursereau, R.; Noel, L.; Brichard, S.M. Skeletal Muscle Secretome in Duchenne Muscular Dystrophy: A Pivotal Anti-Inflammatory Role of Adiponectin. Cell. Mol. Life Sci. 2017, 74, 2487–2501.
- Abou-Samra, M.; Selvais, C.M.; Boursereau, R.; Lecompte, S.; Noel, L.; Brichard, S.M. AdipoRon, a New Therapeutic Prospect for Duchenne Muscular Dystrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 518–533.
- Pillon, N.J.; Krook, A. Innate Immune Receptors in Skeletal Muscle Metabolism. Exp. Cell Res. 2017, 360, 47–54.
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida Albicans. Cell Host Microbe 2009, 5, 487–497.
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.B.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity 2010, 32, 367–378.
- Gimenez, F.; Bhela, S.; Dogra, P.; Harvey, L.; Varanasi, S.K.; Jaggi, U.; Rouse, B.T. The Inflammasome NLRP3 Plays a Protective Role against a Viral Immunopathological Lesion. J. Leukoc. Biol. 2016, 99, 647–657.
- Yang, X.; Zhao, G.; Yan, J.; Xu, R.; Che, C.; Zheng, H.; Zhu, G.; Zhang, J. Pannexin 1 Channels Contribute to IL-1β Expression via NLRP3/Caspase-1 Inflammasome in Keratitis. Curr. Eye Res. 2019, 44, 716–725.
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221.
- Jorquera, G.; Russell, J.; Monsalves-Álvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcón, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254.
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188.
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102.
- Haneklaus, M.; O’Neill, L.A.J. NLRP3 at the Interface of Metabolism and Inflammation. Immunol. Rev. 2015, 265, 53–62.
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Investig. 2006, 116, 3015–3025.
- Barra, N.G.; Henriksbo, B.D.; Anhê, F.F.; Schertzer, J.D. The NLRP3 Inflammasome Regulates Adipose Tissue Metabolism. Biochem. J. 2020, 477, 1089–1107.
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481.
- Field, B.C.; Gordillo, R.; Scherer, P.E. The Role of Ceramides in Diabetes and Cardiovascular Disease Regulation of Ceramides by Adipokines. Front. Endocrinol. 2020, 11, 569250.
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome-Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269.
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and Insulin Resistance. J. Clin. Investig. 2006, 116, 1793–1801.
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646.
- Ogawa, S.; Yakabe, M.; Akishita, M. Age-Related Sarcopenia and Its Pathophysiological Bases. Inflamm. Regen. 2016, 36, 17.
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of Insulin Resistance in Skeletal Muscle. J. Biomed. Biotechnol. 2010, 2010, 476279.
- McCarthy, J.J.; Esser, K.A. Anabolic and Catabolic Pathways Regulating Skeletal Muscle Mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 230–235.
- Riuzzi, F.; Sorci, G.; Arcuri, C.; Giambanco, I.; Bellezza, I.; Minelli, A.; Donato, R. Cellular and Molecular Mechanisms of Sarcopenia: The S100B Perspective. J. Cachexia Sarcopenia Muscle 2018, 9, 1255–1268.
- Salminen, A.; Hyttinen, J.M.T.; Kauppinen, A.; Kaarniranta, K. Context-Dependent Regulation of Autophagy by IKK-NF-κB Signaling: Impact on the Aging Process. Int. J. Cell Biol. 2012, 2012, 849541.
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714.
- Fan, J.; Kou, X.; Yang, Y.; Chen, N. MicroRNA-Regulated Proinflammatory Cytokines in Sarcopenia. Mediat. Inflamm. 2016, 2016, 1438686.
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune-Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590.
- Cordero, M.D.; Williams, M.R.; Ryffel, B. AMP-Activated Protein Kinase Regulation of the NLRP3 Inflammasome during Aging. Trends Endocrinol. Metab. 2018, 29, 8–17.
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532.
- McBride, M.J.; Foley, K.P.; D’Souza, D.M.; Li, Y.E.; Lau, T.C.; Hawke, T.J.; Schertzer, J.D. The NLRP3 Inflammasome Contributes to Sarcopenia and Lower Muscle Glycolytic Potential in Old Mice. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E222–E232.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708.
- Nabavi, S.M.; Nabavi, S.F.; Sureda, A.; Xiao, J.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Baldi, A.; Khan, H.; Daglia, M. Anti-Inflammatory Effects of Melatonin: A Mechanistic Review. Crit. Rev. Food Sci. Nutr. 2019, 59, S4–S16.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; López, L.C.; Mokhtar, D.M.; Acuña-Castroviejo, D. The Protective Effect of Melatonin Against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. Ser. A 2018, 73, 1330–1338.
- Sayed, R.K.; Fernández-Ortiz, M.; Fernández-Martínez, J.; Aranda Martínez, P.; Guerra-Librero, A.; Rodríguez-Santana, C.; de Haro, T.; Escames, G.; Acuña-Castroviejo, D.; Rusanova, I. The Impact of Melatonin and NLRP3 Inflammasome on the Expression of microRNAs in Aged Muscle. Antioxidants 2021, 10, 524.
- Annex, B.H. Therapeutic Angiogenesis for Critical Limb Ischaemia. Nat. Rev. Cardiol. 2013, 10, 387–396.
- Albadawi, H.; Oklu, R.; Cormier, N.R.; O’Keefe, R.M.; Heaton, J.T.; Kobler, J.B.; Austen, W.G.; Watkins, M.T. Hind Limb Ischemia-Reperfusion Injury in Diet-Induced Obese Mice. J. Surg. Res. 2014, 190, 683–691.
- Elshaer, S.; Mohamed, I.; Coucha, M.; Altantawi, S.; Eldahshan, W.; Bartasi, M.; Shanab, A.; Lorys, R.; El-Remessy, A. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017, 6, 47.
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.-H.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme Oxygenase-1 Drives Metaflammation and Insulin Resistance in Mouse and Man. Cell 2014, 158, 25–40.
- Puthucheary, Z.A.; Rawal, J.; McPhail, M.; Connolly, B.; Ratnayake, G.; Chan, P.; Hopkinson, N.S.; Padhke, R.; Dew, T.; Sidhu, P.S.; et al. Acute Skeletal Muscle Wasting in Critical Illness. JAMA 2013, 310, 1591.
- Wollersheim, T.; Woehlecke, J.; Krebs, M.; Hamati, J.; Lodka, D.; Luther-Schroeder, A.; Langhans, C.; Haas, K.; Radtke, T.; Kleber, C.; et al. Dynamics of Myosin Degradation in Intensive Care Unit-Acquired Weakness during Severe Critical Illness. Intensive Care Med. 2014, 40, 528–538.
- Valentine, R.J.; Jefferson, M.A.; Kohut, M.L.; Eo, H. Imoxin Attenuates LPS-Induced Inflammation and MuRF1 Expression in Mouse Skeletal Muscle. Physiol Rep. 2018, 6, e13941.
- Li, R.; Lu, K.; Wang, Y.; Chen, M.; Zhang, F.; Shen, H.; Yao, D.; Gong, K.; Zhang, Z. Triptolide Attenuates Pressure Overload-Induced Myocardial Remodeling in Mice via the Inhibition of NLRP3 Inflammasome Expression. Biochem. Biophys. Res. Commun. 2017, 485, 69–75.
- Fang, W.-Y.; Tseng, Y.-T.; Lee, T.-Y.; Fu, Y.-C.; Chang, W.-H.; Lo, W.-W.; Lin, C.-L.; Lo, Y.-C. Triptolide Prevents LPS-Induced Skeletal Muscle Atrophy via Inhibiting NF-κB/TNF-α and Regulating Protein Synthesis/degradation Pathway. Br. J. Pharmacol. 2021, 178, 2998–3016.
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-Sulfonyl Nitrile Compound, Safe in Humans, Inhibits the NLRP3 Inflammasome and Reverses the Metabolic Cost of Inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539.
- Huang, N.; Kny, M.; Riediger, F.; Busch, K.; Schmidt, S.; Luft, F.C.; Slevogt, H.; Fielitz, J. Deletion of Nlrp3 Protects from Inflammation-Induced Skeletal Muscle Atrophy. Intensive Care Med. Exp. 2017, 5, 3.
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745.
- González-Jamett, A.M.; Bevilacqua, J.A.; Díaz, A.M.C. Hereditary Myopathies. In Muscle Cell and Tissue—Current Status of Research Field; BoD—Books on Demand: Norderstedt, Germany, 2018.
- Chang, L.; Niu, F.; Chen, J.; Cao, X.; Liu, Z.; Bao, X.; Xu, Y. Ghrelin Improves Muscle Function in Dystrophin-Deficient Mdx Mice by Inhibiting NLRP3 Inflammasome Activation. Life Sci. 2019, 232, 116654.
- Aoki, M. LGMD2B (dysferlin deficiency). Ryoikibetsu Shokogun Shirizu 2001, 35, 84–87. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-0035237626&origin=inward&txGid=e48ffe062c960be6c2825a0d55ca7952 (accessed on 12 October 2021).
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842.
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of Adiponectin in the Pathogenesis of Dystrophinopathy. Skelet. Muscle 2015, 5, 25.
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. New Targets to Alleviate Skeletal Muscle Inflammation: Role of microRNAs Regulated by Adiponectin. Sci. Rep. 2017, 7, 43437.
- Dong, X.; Hui, T.; Chen, J.; Yu, Z.; Ren, D.; Zou, S.; Wang, S.; Fei, E.; Jiao, H.; Lai, X. Metformin Increases Sarcolemma Integrity and Ameliorates Neuromuscular Deficits in a Murine Model of Duchenne Muscular Dystrophy. Front. Physiol. 2021, 12, 642908.
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol Induces Expression of the Slow, Oxidative Phenotype in Mdx Mouse Muscle Together with Enhanced Activity of the SIRT1-PGC-1α Axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82.
- Bastin, J.; Djouadi, F. Resveratrol and Myopathy. Nutrients 2016, 8, 254.
- Dial, A.G.; Ng, S.Y.; Manta, A.; Ljubicic, V. The Role of AMPK in Neuromuscular Biology and Disease. Trends Endocrinol. Metab. 2018, 29, 300–312.
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much More than a Hunger Hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624.
- Pan, Y.; Chen, C.; Shen, Y.; Zhu, C.-H.; Wang, G.; Wang, X.-C.; Chen, H.-Q.; Zhu, M.-S. Curcumin Alleviates Dystrophic Muscle Pathology in Mdx Mice. Mol. Cells 2008, 25, 531–537.
- Dubuisson, N.; Abou-Samra, M.; Davis, M.; Noel, L.; Selvais, C.; Brichard, S. DMD—ANIMAL MODELS: EP. 88 Inflammasome Inhibitors for the Treatment of Muscular Dystrophies. Neuromuscul. Disord. 2021, 31, S76.
- Dalakas, M.C. Inflammatory Muscle Diseases. N. Engl. J. Med. 2015, 373, 393–394.
- Dobloug, C.; Garen, T.; Bitter, H.; Stjärne, J.; Stenseth, G.; Grøvle, L.; Sem, M.; Gran, J.T.; Molberg, Ø. Prevalence and Clinical Characteristics of Adult Polymyositis and Dermatomyositis; Data from a Large and Unselected Norwegian Cohort. Ann. Rheum. Dis. 2015, 74, 1551–1556.
- Henriques-Pons, A.; Nagaraju, K. Nonimmune Mechanisms of Muscle Damage in Myositis: Role of the Endoplasmic Reticulum Stress Response and Autophagy in the Disease Pathogenesis. Curr. Opin. Rheumatol. 2009, 21, 581–587.
- Liu, D.; Xiao, Y.; Zhou, B.; Gao, S.; Li, L.; Zhao, L.; Chen, W.; Dai, B.; Li, Q.; Duan, H.; et al. PKM2-Dependent Glycolysis Promotes Skeletal Muscle Cell Pyroptosis by Activating the NLRP3 Inflammasome in Dermatomyositis/polymyositis. Rheumatology 2021, 60, 2177–2189.
- Ma, M.; Chai, K.; Deng, R. Study of the Correlation between the Noncanonical Pathway of Pyroptosis and Idiopathic Inflammatory Myopathy. Int. Immunopharmacol. 2021, 98, 107810.
- Kang, J.; Feng, D.; Yang, F.; Tian, X.; Han, W.; Jia, H. Comparison of Rapamycin and Methylprednisolone for Treating Inflammatory Muscle Disease in a Murine Model of Experimental Autoimmune Myositis. Exp. Ther. Med. 2020, 20, 219–226.
- Lundberg, I.; Kratz, A.K.; Alexanderson, H.; Patarroyo, M. Decreased Expression of Interleukin-1alpha, Interleukin-1beta, and Cell Adhesion Molecules in Muscle Tissue Following Corticosteroid Treatment in Patients with Polymyositis and Dermatomyositis. Arthritis Rheum. 2000, 43, 336–348.
- Tucci, M.; Quatraro, C.; Dammacco, F.; Silvestris, F. Interleukin-18 Overexpression as a Hallmark of the Activity of Autoimmune Inflammatory Myopathies. Clin. Exp. Immunol. 2006, 146, 21–31.
- Yin, X.; Han, G.-C.; Jiang, X.-W.; Shi, Q.; Pu, C.-Q. Increased Expression of the NOD-like Receptor Family, Pyrin Domain Containing 3 Inflammasome in Dermatomyositis and Polymyositis Is a Potential Contributor to Their Pathogenesis. Chin. Med. J. 2016, 129, 1047–1052.
- Chinoy, H.; Li, C.K.-C.; Platt, H.; Fertig, N.; Varsani, H.; Gunawardena, H.; Betteridge, Z.; Oddis, C.V.; McHugh, N.J.; Wedderburn, L.R.; et al. Genetic Association Study of NF-κB Genes in UK Caucasian Adult and Juvenile Onset Idiopathic Inflammatory Myopathy. Rheumatology 2012, 51, 794–799.
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279.
- Li, Y.; Zhou, Y.; Wang, Q. Multiple Values of F-FDG PET/CT in Idiopathic Inflammatory Myopathy. Clin. Rheumatol. 2017, 36, 2297–2305.
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-Dependent Glycolysis Promotes NLRP3 and AIM2 Inflammasome Activation. Nat. Commun. 2016, 7, 13280.
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-Mediated Mechanisms in the Pathoprogression of Amyotrophic Lateral Sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899.
- Debye, B.; Schmülling, L.; Zhou, L.; Rune, G.; Beyer, C.; Johann, S. Neurodegeneration and NLRP3 Inflammasome Expression in the Anterior Thalamus of SOD1(G93A) ALS Mice. Brain Pathol. 2018, 28, 14–27.
- Heitzer, M.; Kaiser, S.; Kanagaratnam, M.; Zendedel, A.; Hartmann, P.; Beyer, C.; Johann, S. Administration of 17β-Estradiol Improves Motoneuron Survival and Down-Regulates Inflammasome Activation in Male SOD1(G93A) ALS Mice. Mol. Neurobiol. 2017, 54, 8429–8443.
- Kadhim, H.; Deltenre, P.; Martin, J.-J.; Sébire, G. In-Situ Expression of Interleukin-18 and Associated Mediators in the Human Brain of sALS Patients: Hypothesis for a Role for Immune-Inflammatory Mechanisms. Med. Hypotheses 2016, 86, 14–17.
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2016, 2016, 5930621.
- Lehmann, S.; Esch, E.; Hartmann, P.; Goswami, A.; Nikolin, S.; Weis, J.; Beyer, C.; Johann, S. Expression Profile of Pattern Recognition Receptors in Skeletal Muscle of SOD1 Amyotrophic Lateral Sclerosis (ALS) Mice and Sporadic ALS Patients. Neuropathol. Appl. Neurobiol. 2018, 44, 606–627.
- Moreno-García, L.; Miana-Mena, F.J.; Moreno-Martínez, L.; de la Torre, M.; Lunetta, C.; Tarlarini, C.; Zaragoza, P.; Calvo, A.C.; Osta, R. Inflammasome in ALS Skeletal Muscle: As a Potential Biomarker. Int. J. Mol. Sci. 2021, 22, 2523.
- Nishimura, M.; Naito, S. Tissue-Specific mRNA Expression Profiles of Human Toll-like Receptors and Related Genes. Biol. Pharm. Bull. 2005, 28, 886–892.
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle Mitochondrial Uncoupling Dismantles Neuromuscular Junction and Triggers Distal Degeneration of Motor Neurons. PLoS ONE 2009, 4, e5390.
- Boyer, J.G.; Ferrier, A.; Kothary, R. More than a Bystander: The Contributions of Intrinsic Skeletal Muscle Defects in Motor Neuron Diseases. Front. Physiol. 2013, 4, 356.
- Manzano, R.; Toivonen, J.M.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; López-Royo, T.; Molina, N.; Zaragoza, P.; Calvo, A.C.; Osta, R. What Skeletal Muscle Has to Say in Amyotrophic Lateral Sclerosis: Implications for Therapy. Br. J. Pharmacol. 2021, 178, 1279–1297.
- Liu, Y.; Gao, X.; Miao, Y.; Wang, Y.; Wang, H.; Cheng, Z.; Wang, X.; Jing, X.; Jia, L.; Dai, L.; et al. NLRP3 Regulates Macrophage M2 Polarization through up-Regulation of IL-4 in Asthma. Biochem. J. 2018, 475, 1995–2008.
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 Acts as a Myoblast Recruitment Factor during Mammalian Muscle Growth. Cell 2003, 113, 483–494.
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The Microglial NLRP3 Inflammasome Is Activated by Amyotrophic Lateral Sclerosis Proteins. Glia 2020, 68, 407–421.
- Coll, R.C.; O’Neill, L.; Schroder, K. Questions and Controversies in Innate Immune Research: What Is the Physiological Role of NLRP3? Cell Death Discov. 2016, 2, 16019.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
30 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No