 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vsevolod V Gurevich | + 3178 word(s) | 3178 | 2021-11-24 07:19:07 | | | |
| 2 | Amina Yu | Meta information modification | 3178 | 2021-11-25 04:03:28 | | |
Video Upload Options
G-protein coupled receptors (GPCRs) are the largest known family of signaling proteins, with over 800 members in humans, and even more in most mammalian species. They are responsible for initiating intracellular signaling that affects metabolism, growth, differentiation, and mediate sensory inputs underlying taste, sense of smell, and vision. GPCRs are targeted by about a third of clinically used drugs.
1. Introduction
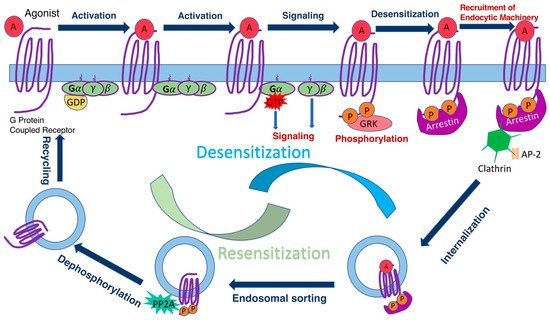
G-protein coupled receptors (GPCRs) are the largest known family of signaling proteins, with over 800 members in humans, and even more in most mammalian species ( http://gpcrdb.org , accessed on 16 on November 2021). They are responsible for initiating intracellular signaling that affects metabolism, growth, differentiation, and mediate sensory inputs underlying taste, sense of smell, and vision [1]. GPCRs are targeted by about a third of clinically used drugs [2]. While there is a lot of structural and dynamic information about how GPCRs engage G-proteins, less is known about how arrestins interact with GPCRs. Arrestins are regulatory proteins that play a key role in homologous desensitization of GPCRs ( Figure 1 ) and direct signaling to other pathways [3][4]. Existing structures of two different arrestins (arrestin-1 and arrestin-2) bound to several GPCRs [5][6][7][8][9][10] reveal the end result, but do not reveal the sequence of molecular events during binding or identify arrestin and receptor elements that drive the process. Receptor binding of arrestins is precisely timed, and arrestins demonstrate impressive selectivity for active phosphorylated receptors [11][12]. Timing and selectivity of arrestin binding was shown to be critical for the proper function of rod photoreceptors in the retina [13], as well as in other GPCR-driven signaling systems [14].
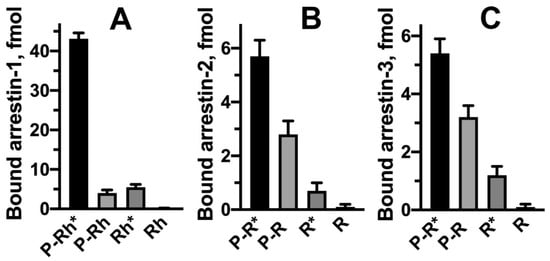
Only four subtypes of arrestin proteins are expressed in most vertebrates (bony fish that underwent an extra round of whole genome duplication express more) [15]. Arrestin-1 and -4 are the two visual subtypes present in photoreceptor cells in the retina. Their function is to regulate phototransduction cascades, i.e., quench photopigment signaling in rods and cones [16]. Arrestin-2 and -3 are ubiquitously expressed and regulate hundreds of GPCR subtypes involved in a wide range of signaling pathways [3][4]. Arrestin-1 displays the highest selectivity for active phosphorylated rhodopsin, as compared to its other functional forms [11], including retinal-free opsin [17]. Therefore, it was used to elucidate the mechanism of arrestin activation and its binding to various functional forms of cognate receptors. Similar to rhodopsin, non-visual GPCRs can be in an active or inactive state, unphosphorylated or phosphorylated. It should be noted, though, that in contrast to rhodopsin, which exists in the dark with covalently attached inverse agonist 11-cis-retinal, effectively suppressing its constitutive activity (reviewed in [18]), non-visual GPCRs in an unliganded state, as well as occupied by various ligands, sample a variety of conformations [19]. Additionally, in rhodopsin, all GRK-phosphorylated residues are compactly localized in a short stretch in the C-terminus [13], while in other receptors, GRK targets can be in the C-terminus or cytoplasmic loops, often in more than one receptor element (e.g., see several examples in [20] and more general discussion in [3]). However, critical mechanisms that govern the arrestin–GPCR interactions appear to be conserved in other arrestin subtypes [11], although their relative roles in the binding vary. Two possible modes of the binding of non-visual arrestins to their cognate GPCRs have been documented: the engagement of only the phosphorylated C-terminus and simultaneous two-site interaction with the C-terminus and the cavity between transmembrane helices [21]. Interestingly, the first mode of binding was shown to be compatible with simultaneous interaction with arrestin and G protein [22]. Even though this was demonstrated using an artificial construct, β2-adrenergic receptor (β2AR) with vasopressin V2 receptor C-terminus, it is entirely possible that this might be applicable to natural non-visual GPCRs. Arrestin-1 demonstrates virtually no binding to unphosphorylated inactive rhodopsin (Rh) or unphosphorylated opsin [17]. Relatively low binding to unphosphorylated, active rhodopsin (Rh*), phosphorylated inactive rhodopsin (P-Rh), and phosphorylated opsin was detected [17]. In fact, phosphorylated opsin is the second most “attractive” functional form of rhodopsin after phosphorylated active rhodopsin (P-Rh*) [17][23]. Arrestin-1 demonstrates 10- to 20-fold higher binding to P-Rh * [17].
This high specificity was explained by the existence of two independent elements (termed sensors [17]) in arrestin-1, one interacting with the receptor-attached phosphates and the other with parts of the receptor that change conformation upon activation [11][17]. The sequential multi-site model of the arrestin–receptor interaction posits that to swing into action, arrestins need both the receptor in active conformation and receptor-attached phosphates to simultaneously engage arrestin elements that specifically bind respective parts of the receptor [11]. In the case of arrestin-1, the only functional form of rhodopsin capable of engaging both sensors at the same time is P-Rh* [17]. This triggers a global conformational change in arrestin, which brings to the receptor additional elements that stabilize this interaction, and therefore increase the binding affinity [11]. This model is consistent with available structures of free (in the basal conformation) [24][25][26][27] and receptor-bound [5][6][7][8][9][10] arrestins, and molecular modeling of arrestin activation [28]. Conformational changes in arrestin-3 (i.e., β−arrestin2) induced by GPCR binding were explored in cells using several sensors incorporated into the arrestin molecule [29][30]. Additionally, differentially phosphorylated rhodopsin [31] and vasopressin receptor C-termini [32] were used to investigate how the pattern of receptor phosphorylation (usually termed “barcode”) affects arrestin binding and binding-induced conformational rearrangements. Nonetheless, the exact mechanism of the conformational change in arrestin induced by GPCR binding has not been fully elucidated. The first step in this direction is the identification of arrestin sensors.
2. Phosphate Sensor
Numerous mutants of arrestin-1, -2, and -3 have been constructed and tested over the years, some of which demonstrated increased binding to P-Rh* and even unphosphorylated Rh*. When tested, these mutants demonstrate greater overall flexibility [33] and greater propensity to spontaneously assume a receptor-bound-like conformation [34]. These mutants in different studies were termed “enhanced”, “pre-activated”, or “active”. Below, we use the term “pre-activated”, indicating their increased binding to cognate receptors. We do not believe the term “active” correctly describes these mutants, as pre-activated mutants of non-visual arrestins do not facilitate ERK1/2 activation without binding to receptors [35][36], indicating that the conformations of even the most pre-activated mutants are distinct from those of receptor-bound arrestins.
It turned out that lariat loop lysine mutations do not have a significant effect on the binding of WT arrestins to P-Rh* and phosphorylated non-visual receptors [37]. In fact, charge reversal and neutralization of the lariat loop lysine on some pre-activated backgrounds in arrestin-1 even increased the binding to Rh*, suggesting that it might be involved in interactions with unphosphorylated portions of rhodopsin. Thus, while the conserved lysine in the lariat loop apparently participates in receptor binding, it does not act as the phosphate sensor [37]. This lysine is not the only lariat loop residue implicated in rhodopsin binding: alanine substitutions of Leu249, Tyr250, Ser251, Ser252, and Tyr254 in the lariat loop were shown to reduce P-Rh* binding [38].
The role of the two lysines in the β-strand I in binding to cognate GPCRs of non-visual arrestin-2 and -3 was tested using the in-cell BRET-based assay [39][40]. The binding of both non-visual subtypes to P-Rh* in vitro and the binding of arrestin-1 to non-visual GPCRs in cells was dramatically reduced by alanine substitutions of these two lysines [40]. These interactions with non-cognate receptors were likely largely driven by the phosphate binding, so that these data support the idea that these two lysines are critical for phosphate-induced arrestin activation. However, the binding of both arrestin-2 and -3 to cognate non-visual GPCRs in cells was not dramatically impacted by the elimination of positive charges in these positions [39][40]. Thus, non-visual arrestins do not appear to rely as much on receptor-attached phosphates as arrestin-1.
Both receptor phosphorylation and activation are needed for high-affinity arrestin binding, although non-visual arrestin-2/3 are less selective than arrestin-1 ( Figure 2 ). Both sensors must be engaged simultaneously to trigger arrestin transition into a high-affinity receptor-binding conformation, as evidenced by direct binding data [17][41][42][43] and suggested by modeling [28]. When arrestin interacts with the active phosphorylated receptor, the phosphate sensor engages receptor-attached phosphates, while the activation sensor binds parts of the receptor that change conformation upon activation, destabilizing inter-domain interactions. This model of arrestin binding suggests functional symmetry between phosphorylation and activation (reviewed in [11]). While the mechanism of phosphate sensing was thoroughly investigated, the identity of the activation sensor is still debated.
3. Finger Loop as an Activation Sensor
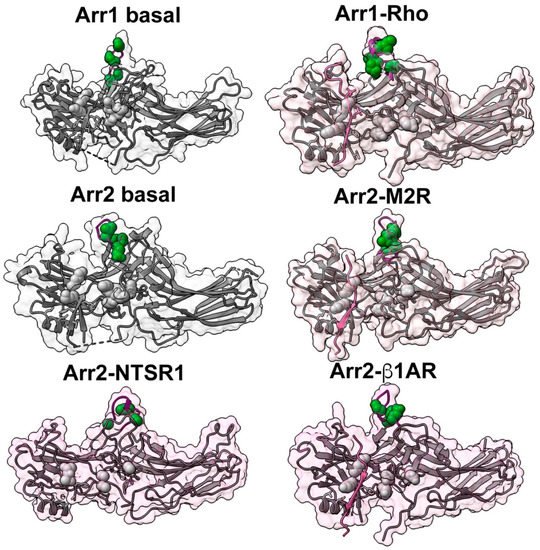
Arrestin finger loop connecting β-strands V and VI emerged as the top candidate for the receptor activation sensor in visual arrestin-1 [44] ( Figure 3 ). The sequence of this loop is remarkably conserved among arrestin subtypes, and the finger loop was invariably found in the cavity between the cytoplasmic ends of GPCR transmembrane helices in all structures of the complex [5][6][7][8][9][10]. As GPCR activation is always accompanied by the opening of this cavity [19][45], it makes perfect sense that the presence of an open cavity serves as an indicator of receptor activation for all proteins that prefer active GPCRs over inactive , G proteins, G protein-coupled receptor kinases, and arrestins [12][46][47]. In the protomers where it is resolved, the finger loop in the basal state of arrestin-1 is unstructured [24][48][49]. This loop forms a short α-helix upon binding rhodopsin [5][6] ( Figure 3 ). While a similar conformational change in the finger loop appears to occur in arrestin-2 bound to the neurotensin receptor 1 [7][10], it was not observed in complex with the muscarinic M2 [8] or with β1-adrenergic [9] receptor ( Figure 3 ). Interestingly, even though the orientation of bound arrestin relative to the receptor in the case of rhodopsin [5][6], M2 receptor [8], β1-adrenergic receptor [9], vasopressin 2 [50], corticotropin-releasing factor receptor 1 [50], and parathyroid hormone receptor 1 [50], on the one hand, and neurotensin receptor [7][10] on the other differs by almost 90°, the finger loop is invariably found within this receptor cavity. It is worth noting that in the arrestin-3 trimer, where all three protomers were in a receptor bound-like conformation, the finger loops of the three protomers engage each other in a manner mimicking their interaction within the inter-helical cavity of GPCRs [51].
Comprehensive alanine-scanning mutagenesis showed that several mutations in the finger loop affect the receptor binding of arrestin-1 [23][38]. Arrestin-3 is the non-visual subtype that interacts with the widest variety of GPCRs [27][52]. Mutations in its finger loop impacted the interactions with receptors, suggesting that it is an essential element in binding. Deletion of the first glycine in the arrestin-3 finger loop drastically reduced binding to several non-visual GPCRs [53]. Proline substitutions that reduce the flexibility and disrupt the helicity of the finger loop also lowered arrestin-3 binding to tested receptors [51]. Consistent with the direct involvement of this element in receptor binding, numerous finger loop residues were found to be strongly immobilized by the bound receptor in arrestin-1 and arrestin-2 [54][55]. Thus, both structural and biochemical evidence is consistent with the idea that the finger loop acts as a sensor of receptor activation.
The finger loop between β-strands V and VI encompasses 11 residues (68–78) in bovine arrestin-1 [24]. It has the same length in bovine arrestin-2 (64–74) [25] and -3 (65–75) [27]. The role of the finger loop in activation recognition was investigated thoroughly by mutagenesis in arrestin-1 [44]. Finger loop mutations were introduced on the background of WT arrestin-1 and truncated arrestin-1-(1-378), which demonstrates much higher binding to Rh* and inactive P-Rh. The binding to Rh*, which lacks receptor-attached phosphates, is mediated only by the activation recognition, so the finger loop substitutions were expected to impact the binding to Rh* more significantly than to P-Rh *. In contrast, dark P-Rh is inactive, so mutations of residues responsible for the activation recognition were expected to minimally affect the binding to this form of rhodopsin.
Both WT and truncated arrestin-1 exhibited the highest binding to P-Rh * [44]. To specifically study the effects on the binding to the active unphosphorylated receptor, the ability of (1–378) mutant to bind to Rh* at relatively high levels was utilized. The binding of truncated arrestin-1 to Rh* was more sensitive to finger loop substitutions than the binding to dark P-Rh and P-Rh *, consistent with the idea this element is an important site for the recognition of the active receptor conformation. Four key residues were identified (Gly-68, Glu-70, and Ile-72 in the finger loop and immediately following Phe-79 in bovine arrestin-1) ( Figure 3 ) for which mutations had significantly reduced the binding to Rh * but not to other functional forms of rhodopsin [44]. Interestingly, these four residues are conserved in non-visual subtypes [15][56] ( Figure 3 ). The mutagenesis data indicate that these four residues are critical for the activation recognition. This idea is consistent with observed crosslinking of arrestin-2 finger loop residues (Asp-67, Leu-68, Val-70, and Leu-71) with the NTSR1 receptor [7].
4. Additional Receptor-Binding Elements
In addition to the finger loop between β-strands V and VI, structural studies have identified a loop in the C-domain (loop XVIII-XIX or loop-344) that determines differential binding to distinct functional forms of the receptor [57]. Conformational changes in the finger loop were directly linked to receptor activation [57], consistent with the idea that it serves as an activation sensor. In contrast, loop-344 was involved in receptor binding in a manner independent of the receptor activation state. Thus, a binding model was proposed in which the finger loop directly engages the activated receptor, whereas loop-344 engages the membrane or neighboring receptor, regardless of the phosphorylation state of the second receptor [57]. This model was supported by the observed role of the arrestin C-domain in phosphorylation-independent low-affinity receptor binding [57]. Molecular dynamics simulation also revealed membrane-touching loops at the distal tip of the C-domain of arrestin that act as membrane anchors, whose engagement is needed to facilitate GPCR binding by arrestins [6].
Structures of arrestin-1 and -2 complexes with several GPCRs revealed other arrestin elements that contact receptors and therefore likely play a role in binding. In the crystal structure of rhodopsin bound to arrestin-1, the phosphorylated rhodopsin–arrestin interface is an intermolecular β-sheet with a network of electrostatic interactions formed between the phosphorylated C-terminus of rhodopsin and the N-terminal domain of arrestin [6]. Phosphorylation was observed at rhodopsin C-terminus residues Thr-336 and Ser-338, which in conjunction with Glu-341 created an extensive network of electrostatic interactions with three positively charged pockets in arrestin-1 [6]. These pockets are formed by three groups of basic residues: Lys-16, Arg-19, and Arg-172 (pocket 1); Lys-16, Arg-30, and Lys-301 (pocket 2); and Lys-15 and Lys-111 (pocket 3), which interact with the rhodopsin C-terminus phosphate groups at Thr-336 and Ser-338, and the negatively charged Glu-341, respectively [6]. The structure with improved resolution [6], as compared to the original one [5], revealed the position of the rhodopsin C-terminus in the complex, which was found to serve as part of the receptor–arrestin interface. The C-terminal portion from Lys-339 to Thr-342 of rhodopsin becomes a β-strand that forms an extended intermolecular antiparallel β-sheet with β-strand I from Val-12 to Lys-16 of the N-terminal domain of arrestin-1 [6]. This rhodopsin β-strand had the same structure in all four complexes in the asymmetric unit, supporting its role as an important part of the arrestin–rhodopsin interface.
The arrestin-2–neurotensin receptor 1 (NTSR1) complex involves intermolecular interactions that include the core interface with two separated patches between the central crest loops of arrestin-2 and the intracellular side of the receptor transmembrane domain, and a tail interface between the arrestin-2 N-domain and the receptor C-terminus [7]. One patch of the core interface is the finger loop. The finger loop makes a direct contact with the turn between TM7 and helix 8 of the receptor [7]. In the arrestin-2-NTSR1 complex, the receptor interacts with arrestin-2 via part of its C-terminus, the transmembrane core, and the C-terminal end of the intracellular loop 3 [10]. Importantly, the C-edge, portions of the 340-loop (residues 330–340) and 191-loop (residues 186–198), seem to be in contact with the detergent micelle. In arrestin-1, homologous 344-loop and 197-loop engage the membrane [58].
Intriguingly, β 1AR [9] and M2R [8] have the same orientation relative to arrestin-2 as rhodopsin relative to arrestin-1 [5][6], whereas NTSR1 is oriented relative to arrestin-2 in a strikingly different manner, rotated by ~90 degrees in both solved structures [7][10]. The finding that the same arrestin can engage different receptors in a distinct fashion suggests that more than one “flavor” of arrestin complex with the same receptor might be possible. In fact, multiple distances were measured by the pulse EPR technique double electron-electron resonance between selected points in rhodopsin and arrestin-1 [5][6]. While the most populated distances matched the structure, the presence of others suggested that different complexes are formed, only one of which is resolved in crystal. The popular hypothesis that the pattern of receptor phosphorylation determines the functional outcome of arrestin binding (barcode hypothesis [59][60]) also implies that arrestin bound to the same differentially phosphorylated receptor can have different conformations.
References
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842.
- Gurevich, V.V.; Gurevich, E.V. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol. Ther. 2006, 110, 465–502.
- Peterson, Y.K.; Luttrell, L.M. The diverse roles of arrestin scaffolds in g protein-coupled receptor signaling. Pharmacol. Rev. 2017, 69, 256–297.
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature 2015, 523, 561–567.
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of phosphorylation codes for arrestin recruitment by g protein-coupled receptors. Cell 2017, 170, 457–469.
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 2019, 29, 971–983.
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302.
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β(1)-adrenoceptor. Nature 2020, 583, 862–866.
- Huang, W.; Masureel, M.; Qianhui, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin. Nature 2020, 579, 303–308.
- Gurevich, V.V.; Gurevich, E.V. The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 2004, 25, 105–111.
- Seyedabadi, M.; Gharghabi, M.; Gurevich, E.V.; Gurevich, V.V. Receptor-arrestin interactions: The GPCR perspective. Biomolecules 2021, 11, 218.
- Mendez, A.; Burns, M.E.; Roca, A.; Lem, J.; Wu, L.W.; Simon, M.I.; Baylor, D.A.; Chen, J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 2000, 28, 153–164.
- Carman, C.V.; Benovic, J.L. G-protein-coupled receptors: Turn-ons and turn-offs. Curr. Opin. Neurobiol. 1998, 8, 335–344.
- Indrischek, H.; Prohaska, S.J.; Gurevich, V.V.; Gurevich, E.V.; Stadler, P.F. Uncovering missing pieces: Duplication and deletion history of arrestins in deuterostomes. BMC Evol. Biol. 2017, 17, 163.
- Gurevich, V.V.; Hanson, S.M.; Song, X.; Vishnivetskiy, S.A.; Gurevich, E.V. The functional cycle of visual arrestins in photoreceptor cells. Prog. Retin. Eye Res. 2011, 30, 405–430.
- Gurevich, V.V.; Benovic, J.L. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J. Biol. Chem. 1993, 268, 11628–11638.
- Lamb, T.D. Evolution of phototransduction, vertebrate photoreceptors and retina. Prog. Retin. Eye Res. 2013, 36, 52–119.
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 2015, 161, 1101–1111.
- Gurevich, E.V.; Gurevich, V.V. GRKs as modulators of neurotransmitter receptors. Cells 2020, 10, 52.
- Cahill, T.J., III; Thomsen, A.R.; Tarrasch, J.T.; Plouffe, B.; Nguyen, A.H.; Yang, F.; Huang, L.Y.; Kahsai, A.W.; Bassoni, D.L.; Gavino, B.J.; et al. Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567.
- Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J., III; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G protein-β-arrestin super-complex mediates sustained g protein signaling. Cell 2016, 166, 907–919.
- Peterhans, C.; Lally, C.C.; Ostermaier, M.K.; Sommer, M.E.; Standfuss, J. Functional map of arrestin binding to phosphorylated opsin, with and without agonist. Sci. Rep. 2016, 6, 28686.
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. A Model for Arrestin’s regulation: The 2.8 Å crystal structure of visual arrestin. Cell 1999, 97, 257–269.
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of beta-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane translocation. Structure 2001, 9, 869–880.
- Sutton, R.B.; Vishnivetskiy, S.A.; Robert, J.; Hanson, S.M.; Raman, D.; Knox, B.E.; Kono, M.; Navarro, J.; Gurevich, V.V. Crystal structure of cone arrestin at 2.3Å: Evolution of receptor specificity. J. Mol. Biol. 2005, 354, 1069–1080.
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J. Mol. Biol. 2011, 406, 467–478.
- Sente, A.; Peer, R.; Srivastava, A.; Baidya, M.; Lesk, A.M.; Balaji, S.; Shukla, A.K.; Babu, M.M.; Flock, T. Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 538–545.
- Lee, M.H.; Appleton, K.M.; Strungs, E.G.; Kwon, J.Y.; Morinelli, T.A.; Peterson, Y.K.; Laporte, S.A.; Luttrell, L.M. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 2016, 531, 665–668.
- Nuber, S.; Zabel, U.; Lorenz, K.; Nuber, A.; Milligan, G.; Tobin, A.B.; Lohse, M.J.; Hoffmann, C. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 2016, 531, 661–664.
- Mayer, D.; Damberger, F.F.; Samarasimhareddy, M.; Feldmueller, M.; Vuckovic, Z.; Flock, T.; Bauer, B.; Mutt, E.; Zosel, F.; Allain, F.H.T.; et al. Distinct G protein-coupled receptor phosphorylation motifs modulate arrestin affinity and activation and global conformation. Nat. Commun. 2019, 10, 1261.
- Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; Heydenreich, F.M.; Suomivuori, C.M.; Brinton, C.; Townshend, R.J.L.; Bouvier, M.; Kobilka, B.K.; Dror, R.O. How GPCR phosphorylation patterns orchestrate arrestin-mediated signaling. Cell 2020, 183, 1813–1825.
- Carter, J.M.; Gurevich, V.V.; Prossnitz, E.R.; Engen, J.R. Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J. Mol. Biol. 2005, 351, 865–878.
- Kim, M.; Vishnivetskiy, S.A.; Van Eps, N.; Alexander, N.S.; Cleghorn, W.M.; Zhan, X.; Hanson, S.M.; Morizumi, T.; Ernst, O.P.; Meiler, J.; et al. Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. USA 2012, 109, 18407–18412.
- Coffa, S.; Breitman, M.; Hanson, S.M.; Callaway, K.; Kook, S.; Dalby, K.N.; Gurevich, V.V. The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS ONE 2011, 6, e28723.
- Coffa, S.; Breitman, M.; Spiller, B.W.; Gurevich, V.V. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry 2011, 50, 6951–6958.
- Vishnivetskiy, S.A.; Zheng, C.; May, M.B.; Karnam, P.C.; Gurevich, E.V.; Gurevich, V.V. Lysine in the lariat loop of arrestins does not serve as phosphate sensor. J. Neurochem. 2021, 156, 405–444.
- Ostermaier, M.K.; Peterhans, C.; Jaussi, R.; Deupi, X.; Standfuss, J. Functional map of arrestin-1 at single amino acid resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 1825–1830.
- Gimenez, L.E.; Babilon, S.; Wanka, L.; Beck-Sickinger, A.G.; Gurevich, V.V. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell. Signal. 2014, 26, 1523–1531.
- Gimenez, L.E.; Kook, S.; Vishnivetskiy, S.A.; Ahmed, M.R.; Gurevich, E.V.; Gurevich, V.V. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J. Biol. Chem. 2012, 287, 9028–9040.
- Kovoor, A.; Celver, J.; Abdryashitov, R.I.; Chavkin, C.; Gurevich, V.V. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999, 274, 6831–6834.
- Celver, J.; Vishnivetskiy, S.A.; Chavkin, C.; Gurevich, V.V. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 2002, 277, 9043–9048.
- Gurevich, V.V.; Dion, S.B.; Onorato, J.J.; Ptasienski, J.; Kim, C.M.; Sterne-Marr, R.; Hosey, M.M.; Benovic, J.L. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 1995, 270, 720–731.
- Vishnivetskiy, S.A.; Huh, E.K.; Gurevich, E.V.; Gurevich, V.V. The finger loop as an activation sensor in arrestin. J. Neurochem. 2021, 157, 1138–1152.
- Farrens, D.L.; Altenbach, C.; Yang, K.; Hubbell, W.L.; Khorana, H.G. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 1996, 274, 768–770.
- Gurevich, V.V.; Gurevich, E.V. GPCRs and signal transducers: Interaction stoichiometry. Trends Pharmacol. Sci. 2018, 39, 672–684.
- Chen, Q.; Plasencia, M.; Li, Z.; Mukherjee, S.; Patra, D.; Chen, C.L.; Klose, T.; Yao, X.Q.; Kossiakoff, A.A.; Chang, L.; et al. Structures of rhodopsin in complex with G-protein-coupled receptor kinase. Nature 2021, 595, 600–605.
- Granzin, J.; Wilden, U.; Choe, H.W.; Labahn, J.; Krafft, B.; Buldt, G. X-ray crystal structure of arrestin from bovine rod outer segments. Nature 1998, 391, 918–921.
- Sander, C.L.; Luu, J.; Kim, K.; Furkert, D.; Jang, K.; Reichenwallner, J.; Kang, M.; Lee, H.J.; Eger, B.T.; Choe, H.W.; et al. Structural evidence for visual arrestin priming via complexation of phosphoinositols. Structure 2021, S0969–2126, 00370–00371.
- Böttke, T.; Ernicke, S.; Serfling, R.; Ihling, C.; Burda, E.; Gurevich, V.V.; Sinz, A.; Coin, I. Exploring GPCR-arrestin interfaces with genetically encoded crosslinkers. EMBO Rep. 2020, 21, e50437.
- Chen, Q.; Perry, N.A.; Vishnivetskiy, S.A.; Berndt, S.; Gilbert, N.C.; Zhuo, Y.; Singh, P.K.; Tholen, J.; Ohi, M.D.; Gurevich, E.V.; et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 2017, 8, 1427.
- Barak, L.S.; Ferguson, S.S.; Zhang, J.; Caron, M.G. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 1997, 272, 27497–27500.
- Zheng, C.; Tholen, J.; Gurevich, V.V. Critical role of the finger loop in arrestin binding to the receptors. PLoS ONE 2019, 14, e0213792.
- Hanson, S.M.; Francis, D.J.; Vishnivetskiy, S.A.; Kolobova, E.A.; Hubbell, W.L.; Klug, C.S.; Gurevich, V.V. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. USA 2006, 103, 4900–4905.
- Vishnivetskiy, S.A.; Gimenez, L.E.; Francis, D.J.; Hanson, S.M.; Hubbell, W.L.; Klug, C.S.; Gurevich, V.V. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J. Biol. Chem. 2011, 286, 24288–24299.
- Gurevich, E.V.; Gurevich, V.V. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 236.
- Sommer, M.E.; Hofmann, K.P.; Heck, M. Distinct loops in arrestin differentially regulate ligand binding within the GPCR opsin. Nat. Commun. 2012, 3, 995.
- Lally, C.C.; Bauer, B.; Selent, J.; Sommer, M.E. C-edge loops of arrestin function as a membrane anchor. Nat. Commun. 2017, 8, 14258.
- Nobles, K.N.; Xiao, K.; et Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the 2-adrenergic receptor establish a barcode that encodes differential functions of -arrestin. Sci. Signal. 2011, 4, ra51.
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149.


