

5. AMD Pathogenesis
While the exact pathogenesis of AMD is not fully understood, RPE dysfunction has a crucial role in both dry and wet AMD.
5.1. Complement Dysregulation in AMD
The complement system of innate immunity is essential for preventing inflammation. The eye is an immune-privileged organ, which can tolerate the introduction of antigens with its limited immune responses. The RPE is the primary driver source of complement activation in the retina. The constituents of the complement system are strictly regulated to small quantities in the eye [60]. This system could be activated via classical, mannose-binding lectin, and the alternative pathway. Among these systems, the AP is the major pathway related to AMD pathogenesis.
Inappropriate increases in complement activation are implicated in AMD pathogenesis [60][61][62][63]. Immunocytochemical analysis of drusen components and AMD lesions revealed a significant number of complement components, such as C3, C5, C9, complement factor F and H (CFF, CFH), and membrane attack complex (MAC) [64][65]. The AMD patients have elevated level of C3, C3d, Bb, and C5a [66].
The geographic atrophy (GA) is believed to occur usually because of drusen disturbing the transportation and removal of nutrients and wastes, respectively. This disturbance results in cell death in GA [61]. While the relationship between neovascular AMD and the impaired complement system is unknown, C3a, C5a, complement factor B, and MAC were shown to increase CNV lesions in a laser-induced CNV animal model by increasing the angiogenic factors secretion such as VEGF, TGF-β2, and β-FGF from the RPE [67][68].
The AMD is also affected via genetic variants of the complement system [69][70][71]. Genetic variations in CFB, C2, serpin peptidase inhibitor clade G member 1 (a complement component 1 inhibitor), and C3 increase the risk of AMD. The complement system may exacerbate the chronic local inflammation in AMD. C3a and C5a can stimulate the secretion of inflammatory cytokines including interleukin-1β, -6, -8, granulocyte-macrophage colony-stimulating factor, and MCP-1 from the RPE [72].
Oxidative stress could make the RPE more susceptible to complement-associated injury [73][74]. RPE cells under oxidative stress exhibit reduced expression of CD55 and CD59 and increased expression of CFH [75]. In human RPE cells, VEGF secretion is increased up to 100 times due to synergy between the complement cascade and oxidative stress [76].
5.2. Dysfunctional Mitochondria in RPE
Dysfunctional mitochondria in the RPE may be a critical cause of AMD pathogenesis [77][78][79]. Mitochondria mainly fulfill the demands of energy from cells by producing adenosine triphosphate (ATP) via oxidative phosphorylation, citric acid cycle, and β-oxidation. RPE also metabolizes fatty acids to synthesize β-hydroxybutyrate as an auxiliary source of energy [80]. RPE has abundant mitochondria to fulfill the energy needs for the outer retina [81].
Mitochondrial dysfunction leads to damaged respiration, which results in ROS accumulation [82]. Oxidative stress in mitochondria may aggravate the production of ROS leading to apoptosis. The study showed the mitochondrial-based AMD model via treatment of H2O2 to RPE for exhibiting mitochondrial DNA damage [83]. ROS overproduction could cause and result in significant mitochondrial DNA damage [84][85]. The intracellular ROS level is regulated by the antioxidant system, but an overwhelming ROS level leads to cell damage [86]. Although the mechanism of production of ROS in cells is not exactly clear, theories include cytochrome c interaction, damage to the SOD2 gene, and lipofuscin deposition [87][88][89].
SOD2, a primary antioxidant enzyme, protects the cell from damage due to oxidative stress by removing ROS [90]. In an SOD2-knockout mouse model, an increase in ROS level resulted in mitochondrial changes and RPE dysfunction [91]. ROS levels can increase due to interactions between the mitochondria and cytochrome c oxidase and the accumulations of lipofuscin in RPE cells [92][93].
5.3. Pathways of RPE Cell Death in AMD
The death of photoreceptors and RPE occurs through apoptosis in AMD. However, necrosis and pyroptosis pathways have also been reported [94][95]. In the subsequent sections, we have concisely reviewed necrosis, apoptosis, and pyroptosis, the cell death pathways, to understand the mechanisms of RPE degeneration in AMD.
Necrosis is uncontrolled cell death induced by hypoxia or inflammation. This pathway is activated by an increase in diverse pro-inflammatory proteins, such as nuclear factor-κB, leading to the destruction of the cell membrane. The cell contents spilled in the pericellular space cause inflammation and tissue damage. The binding of the tumor necrosis factor (TNF) ligand to death receptors of cellular membrane and its trimerization initiates the necrosis pathway. This pathway is mostly controlled by receptor-interacting protein kinases (RIPs). Necrosome formation occurs after autophosphorylation of RIPK1 and RIPK3 in the absence of caspase 8. The necrosome phosphorylates mixed lineage kinase domain-like and recruits phosphoglycerate mutase 5, which attaches to the mitochondrial membrane to stimulate dynamin-related protein 1, resulting in death of the cell [94]. The involvement of the necrosis pathway in RPE cell death has been studied previously [96][97]. In these studies, the typical characteristics of necrosis included depletion of ATP and aggregation of RIP3 in ARPE 19 cells treated with H2O2 or tertbutyl hydroperoxide to induce oxidative stress. RPE cell death due to oxidative stress was prevented when RIPK3 was not activated [96]. Furthermore, RPE cells exposed to oxidative stress presented morphological features similar to necrosis such as swelling of the cells and loss of cell membrane integrity [97].
Apoptosis is the process including, inhibition of growing and dividing, eventually resulting in controlled death without leakage of its contents into the nearby environment. It is also called programmed cell death. The activation of a chain of cysteine-aspartic proteases known as caspases initiates the apoptosis. There are two types of caspases: initiator caspases and executioner caspases [98]. The damage of cell activates the initiator caspases (caspases 8 and 9) to induce the activation of executioner caspases (caspases 3, 6, and 7). This process results in DNA and nuclear fragmentation, cytoskeleton destruction, and the formation of apoptotic bodies. Apoptosis can be initiated through the intrinsic and extrinsic pathways. The intrinsic pathway involved in the mitochondria depends on factors secreted from the mitochondria. Stressors such as hypoxia, toxins, radiation, ROS, and viruses activate the intrinsic pathway [98]. The cellular damage leads to severe damage in DNA, which results in the suppression of anti-apoptotic factors and secretion of proapoptotic factors, like Bcl-2-associated X protein (Bax). Under Bax stimulation, the cytochrome C is released into the cytoplasm. Then, the apoptosome is formed via binding of cytochrome c with apoptotic protease activating factor 1 (Apaf1). The apoptosome then induce the activation of pro-caspase-9 and caspase-9. Among them, the caspase-9 induces the apoptosis by activating caspase-3 [98][99]. After binding of the TNF ligand to death receptors, the extrinsic pathway is activated. The death-inducing signaling complex is formed via activation of TNF-receptor 1-associated death domain and Fas-associated death domain. Then, the caspase-8 is activated to induce caspase-3 based apoptosis. The involvement of apoptosis in RPE degeneration is well known. Active caspase-3 was found in the RPE cells of a patient with GA [100]. Furthermore, Ho et al. reported that because c-Jun N-terminal kinases and p38 mitogen-activated protein kinase are necessary for Bax translocation to mitochondria, inhibitors of these suppressed the transposition of Bax to the mitochondria in oxidative stress [101]. In a recent study, exposure of RPE cells to the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which initiates inflammatory cell death, activated the apoptosis and pyroptosis pathways [102].
Pyroptosis is the process of programmed cell death with collateral damage through inflammation. This pathway can be activated by caspase 1 either independently or dependently. In a caspase 1-dependent pathway, inflammasomes play an important role. Inflammasomes are large multiprotein complexes formed in response to pathogen-associated molecular patterns and damage-associated molecular patterns. NOD-like receptor family proteins such as NLPR1, NLPR3, and NLPR4 of inflammasomes activate caspase-1 with ASC as the adapter protein. Caspase-1 then promotes the cleavage of the pro-pyroptotic factor gasdermin D, producing an N-terminal fragment that induces cell death. In case of the caspase-1-independent pathway, caspase-4/5 of human and caspase-11 of mouse promote the cleavage of gasdermin D to activate pyroptosis [95].
NLRP3 inflammasomes have been reported in GA and CNV lesions [103]. The proteolytic cleavage of caspase-3 (apoptotic pathway) and gasdermin D (pyroptotic pathway) was found in the RPE and choroid tissues of rats treated by intravitreal amyloid-beta injections [104]. Exposure of primary RPE cells to oxidative stress primed inflammasome formation, and the mechanism of the cell death was switched from apoptosis to pyroptosis [105]. There are some conflicting reports about the mechanisms of RPE cell death in AMD. Further research is required to better understand of these pathways.
5.4. Autophagy
Autophagy maintains cellular homeostasis by the lysosomal degradation of unused and damaged cellular components [106]. AMPK and mammalian target of rapamycin (mTOR) regulate autophagy as a promotor and an inhibitor, respectively. Autophagy is initiated by the formation of a phagophore from the endoplasmic reticulum. Phagophores stretch and enclose the cytoplasm and organelles to form a double-membrane autophagosome. Several autophagy-related proteins (ATGs) together with the LC3 conjugation system form mature autophagosomes with a closed bilayer membrane. After maturation, the autophagosome fuse with the lysosome to develop the autolysosome, which degrades waste material [106][107]. Autophagy in RPE cells usually occurs to maintain the homeostasis of RPE cells [108].
Although the exact role of autophagy in AMD remains unclear, impaired lysosomal degradation owing to the accumulation of lipofuscin is closely related to autophagy in AMD. Cathepsins are lysosomal proteases in the RPE. They degrade POS, forming lipid peroxidation end products and oxidized low-density lipoproteins in RPE cells [107]. Their accumulation induces RPE stress and activates lipofuscinogenesis [109]. Lipofuscin cannot be broken down by lysosomal enzymes and may augment oxidative stress in the RPE. It can decrease lysosomal cathepsin activity, which could result in the accumulation of autolysosomes, causing drusen.
Markers of autophagy have been discovered in the drusen of AMD donor tissue [110]. A decrease in autophagy occurs when RPE cells are chronically exposed to oxidative stress mediated by H2O2, but autophagic biomarkers increase when RPE cells are exposed to acute oxidative stress. Autophagy is generally activated in early AMD because of compensatory mechanisms increasing oxidative stress in the RPE; however, in the late stages of AMD, the autophagy pathway is unable to counter the large amount of damaged organelles and thus becomes dysfunctional [108]. Potential therapeutic strategies targeting autophagy may be useful. Therefore, further research into this pathway is warranted.
5.5. α. B Crystallins and RPE Crystallin in AMD
αA and αB crystallins are the main members of the small heat shock proteins (sHSP) family. sHSP help assemble cellular proteins, guide misfolded proteins, and prevent proteins from denaturing under external stress [111]. sHSP are involved in anti-apoptotic and proapoptotic pathways. αA crystallin is located largely in photoreceptors, astrocytes, and Müller cells, whereas αB crystallin is located predominantly in the RPE and localized to the mitochondria and Golgi apparatus [112].
In one study, the expression of αB crystallin was significantly increased in the RPE of patients with advanced AMD and drusen of patients with neovascular AMD and early atrophic AMD [113]. The expression of αB crystallin was also increased when RPE cells were exposed to oxidative stress induced by H2O2 [111]. αB crystallin stimulates VEGF and protects the protein against aggregation and unfolding [114]. This is possibly the cause of neovascular AMD developed through VEGF overproduction in the RPE. Other studies have also reported increased αB crystallin expression in angiogenesis and a significant reduction in VEGFA expression in αB crystallin-knockout mice [115][116].
Regarding the protective function of αB crystallin in the retina, αB crystallin prevents oxidative stress-mediated apoptotic cell death of the RPE. An increase in apoptotic activity was reported in a crystallin-knockout mouse model [117]. αB crystallin could potentially inhibit apoptosis via interaction with p53 to prevent its translocation to the mitochondria [118]. The increased production of αB crystallin also inhibits ROS activation to prevent apoptosis [119].
6. Treatment Targeting RPE in AMD
Patients with wet AMD are currently treated with anti-VEGF agents to achieve symptomatic relief. However, dry AMD has limited treatment options, such as lifestyle changes and vitamin supplements. Therapeutic strategies targeting RPE cells include the use of inhibitors of the complement pathway and visual cycle, neurotrophic factors, modulators of lipid metabolism, photobiomodulation (PBM) agents, and cell-based therapy. In the subsequent sections, we concisely review PBM and RPE transplantation methods targeting RPE in AMD.
6.1. PBM
PBM uses radiation in the visible to near-infrared spectrum (500–1000 nm) produced by laser or light-emitting diodes. Near-infrared radiation activates cellular functions by stimulating photoreceptors [120][121]. The therapeutic effect of PBM has been reported in animal models and patients with various retinal diseases, such as AMD [122][123], retinitis pigmentosa, and diabetic retinopathy [124].
6.1.1. Mechanism of PBM
PBM targets the mitochondrial cytochrome oxidase C, which controls the oxygen and nitrite levels in tissues by directly activating mitochondrial respiration and indirectly increasing nitric oxide dissociation. A resultant increase in ATP, cAMP, ROS, and intracellular calcium levels promote anti-inflammation, antioxidation, protein synthesis, anti-apoptosis, and cellular metabolism. PBM inhibits oxidative stress, which increases RPE phagocytosis through the upregulation of phosphorylated Mer tyrosine kinase (MerTK) [125] (Figure 2).
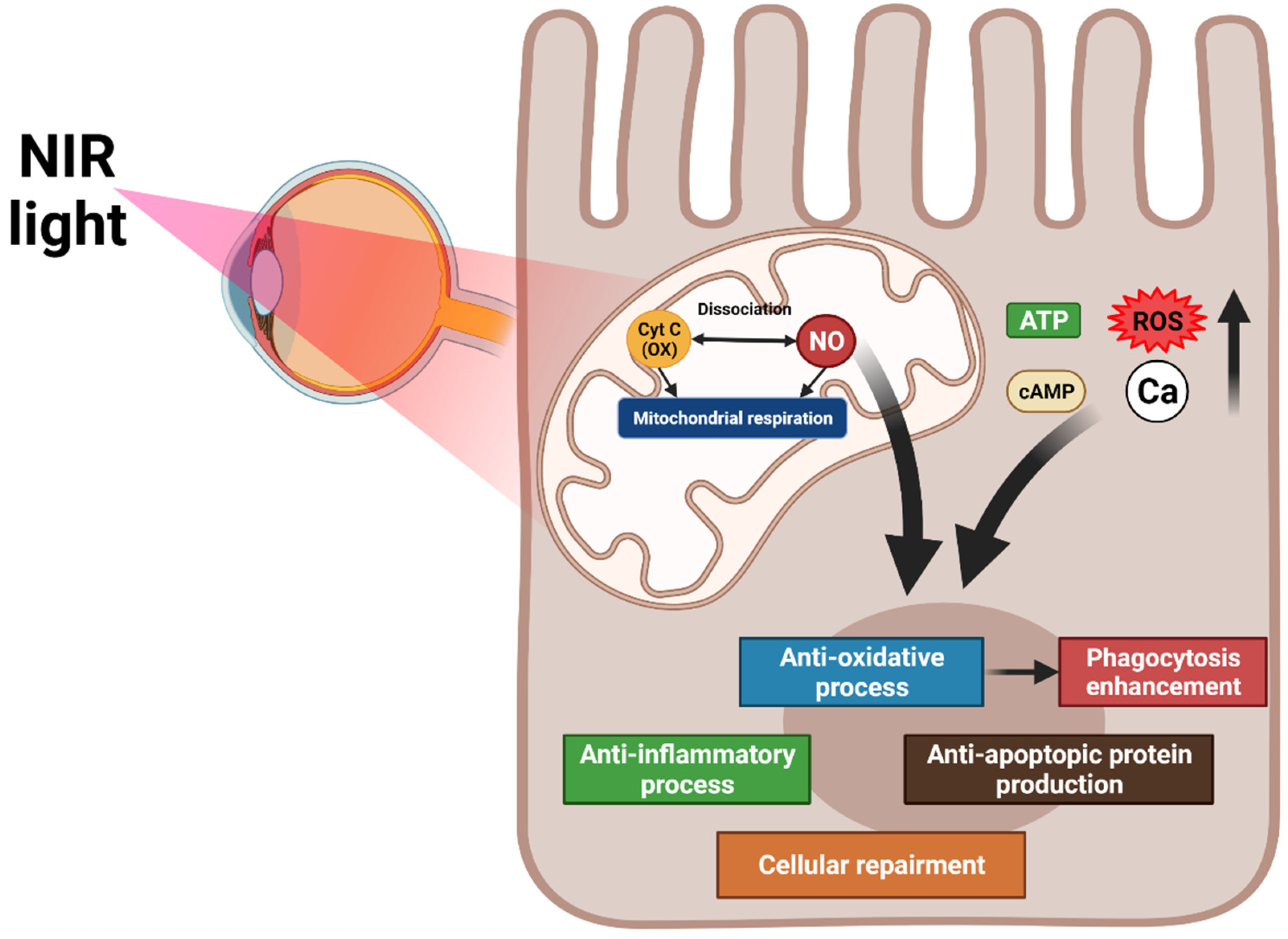
Figure 2. Overview of the mechanisms of photobiomodulation (PBM). PBM activates mitochondrial cytochrome oxidase C and increases mitochondrial respiration and nitric oxide dissociation. These processes elevate ATP, cAMP, reactive oxygen species, and intracellular calcium levels, promoting anti-inflammation, antioxidation, protein synthesis, anti-apoptosis, and cellular metabolism. The antioxidation effect of PBM increases RPE phagocytosis.
6.1.2. Application of PBM at AMD
PBM prevents AMD pathogenesis through its effect on cellular oxidative stress, apoptosis, and inflammation. Furthermore, PBM removes drusen in dry AMD by increasing RPE phagocytosis. Lavey reported that near-infrared radiation increases ATP and nitric oxide levels in RPE cells, which presumably promotes mitochondrial oxidative phosphorylation through the non-binding of nitric oxide from cytochrome oxidase C [126]. Kokkinopoulos et al. reported that after near-infrared radiation light exposure increases the mitochondrial membrane potential and decreases C3d, TNF-α, and macrophages in an animal AMD model, demonstrating an improvement in mitochondrial function and reduction in RPE inflammation [127].
In one study, 348 eyes with dry and wet type AMD were treated with radiation from a 780 nm semiconductor laser diode, and 97% of patients with cataracts and 94% of patients without cataracts showed an average visual improvement and reductions in pigmentation and cystic drusen and improvements in metamorphopsia and dyschromatopsia were observed [122].
The TORPA II study demonstrated functional changes, such as enhancement of contrast sensitivity and visual acuity, and anatomical improvements, such as reduction in drusen volume and central drusen thickness, after PBM in patients with dry AMD [128].
6.2. RPE Cell Transplantation
Unlike for early AMD and wet AMD, no appropriate treatment exists for GA. However, cell-based therapy has recently been used to treat vision loss in late AMD, especially GA. Cell transplants might be used as a rescue or replacement therapy. Rescue therapy may preserve the function of dying tissue and also restore the function of dying cells. Replacement therapy involves the replacement of dead or dying cells with normal cells for restoring the function of a tissue or organ.
Several clinical trials of RPE transplantation for late AMD have demonstrated that improvement in visual function depends on the severity of the damage before RPE cell transplantation. The subsequent sections summarize current clinical trials of RPE transplantation.
6.2.1. RPE Transplantation for Choroidal Neovascularization in AMD
Tezel et al. transplanted allogeneic RPE sheets obtained from cadavers after removing CNV lesions at the subfovea in AMD patients [129]. The patients received RPE cells from different cadavers, and no significant recovery in best-corrected visual acuity and contrast sensitivity was observed. Binder et al. transplanted RPE cells by subretinal injection of autologous RPE suspension after CNV excision at the subfovea in a prospective controlled trial [130]. One year after surgery, the best-corrected visual acuity and reading speed improved. Lu et al. also transplanted autologous RPE sheets in AMD patients after CNV excision [131] and reported recovery in best-corrected visual acuity. Kamao et al. and Nakagawa et al. transplanted autologous induced pluripotent stem cell-derived RPE sheets into the subretinal space in AMD patients after the surgical removal of subfoveal CNV [132][133][134]. Although the sheets were intact one year after surgery, no improvement was noted in the best-corrected visual acuity. Da Cruz et al. transplanted human embryonic stem cell-derived RPE in two AMD patients with subfoveal CNVs. One year after surgery, the best-corrected visual acuity and reading speed improved in only the patient with the least focal foveal atrophy [135]. Recently, the induced pluripotent stem cell (IPSc) based RPE is studied for transplantation [136][137][138]. It is expected to free from immune rejection, the most common adverse effects of transplantation, and showed well attached with AMD patients. However, their visual acuity was nearly unchanged [134].
6.2.2. RPE Transplantation for GA in AMD
Schwartz et al. transplanted embryonic stem cell-derived RPE suspensions in nine AMD patients with GA [139][140]. The improvement in best-corrected visual acuity was 14 letters in eight eyes. Kashani et al. transplanted human embryonic stem cell-derived RPE monolayer sheets (3.5 mm × 6.25 mm) into the GA lesions in five AMD patients [141]. Four of the five patients presented no improvement in vision. However, the vision of the remaining patient improved by 17 ETDRS letters.
7. Conclusions
Because AMD is a multifactorial disease with several pathways, a multifaceted intervention is required. Although the exact pathogenesis of AMD remains unknown, RPE dysfunction is a major contributor. Thus, several therapeutic strategies targeting RPE cells have been proposed. Among these, PBM is a promising noninvasive therapy to ameliorate oxidative stress, mitochondrial dysfunction, and complement dysfunction which are the main mechanisms of RPE dysfunction resulting in AMD. PBM can also remove drusen in AMD by activating RPE phagocytosis. Cell-based therapies involving transplantation of RPE sheets or cells are possible but will likely be beneficial only to patients with minimal photoreceptor atrophy in late AMD. Further research on the relationship between AMD pathogenesis and RPE dysfunction will identify other potential therapeutic targets.


