Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lewis Z. Shi | + 1913 word(s) | 1913 | 2021-11-17 06:38:43 | | | |
| 2 | Nora Tang | Meta information modification | 1913 | 2021-11-23 10:19:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shi, L. The IFN–JAK–STAT AXIS in Radiotherapy and Immunotherapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/16169 (accessed on 08 February 2026).
Shi L. The IFN–JAK–STAT AXIS in Radiotherapy and Immunotherapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/16169. Accessed February 08, 2026.
Shi, Lewis. "The IFN–JAK–STAT AXIS in Radiotherapy and Immunotherapy" Encyclopedia, https://encyclopedia.pub/entry/16169 (accessed February 08, 2026).
Shi, L. (2021, November 18). The IFN–JAK–STAT AXIS in Radiotherapy and Immunotherapy. In Encyclopedia. https://encyclopedia.pub/entry/16169
Shi, Lewis. "The IFN–JAK–STAT AXIS in Radiotherapy and Immunotherapy." Encyclopedia. Web. 18 November, 2021.
Copy Citation
The JAK-STAT pathway is a rapid membrane-to-nucleus signaling module regulated by a wide array of extracellular signals including cytokines and growth factors, as well as cell-intrinsic mutations/alterations. Among all those upstream signals, interferons (IFNs), especially IFN-α/β (belonging to type I IFNs: IFN-Is) and IFN-γ (the only member in type II IFN), are the most widely studied. With their pleiotropic immunological activities in almost all known pathophysiological settings, we discussed the role of the IFN-JAK-STAT axis in radiotherapy (RT) and immunotherapies (IOs), two major pillars of cancer care.
radiotherapy
immunotherapy
interferon
IFN-γ
JAK
STAT
1. The IFN–JAK–STAT Axis in Immunomodulatory Effects of RT
RT can induce double-stranded DNA breaks (DSDBs), small non-coding RNAs (sncRNAs), and danger-associated molecular patterns (DAMPs). While DSDBs primarily activate the cGAS-STING pathway [1] and sncRNAs engage the RIG-I-MAVS pathway [2], different DAMPs are recognized by the host through different mechanisms. For example, translocated calreticulin (CRT) to the outer cell membrane (typically residing in the endoplasmic reticulum) promotes the phagocytosis of irradiated cells by macrophages and dendritic cells (DCs); HMGB1, a highly conserved nonhistone DNA-binding protein, acts as an agonist of Toll-like receptor 2 (TLR2) and TLR4 [3], two primary pattern recognition receptors (PRRs) on DCs; and extracellular ATPs bind to P2X7 purinergic receptors on DC [4][5]. All these danger/stress-associated signals converge on DC activation and maturation, leading to activation of tumor antigen-specific T cells. Two early studies showed that the IFNAR1 expression in DC (but not tumor cells) plays a dominant role in orchestrating the immunological effects of RT. However, it is noteworthy to point out that both studies employed a single high dose of radiation (20 Gy) [6][7], which may not be ideal to induce immunogenic effects. To this end, a recent study reported that single high-dose RT (20 or 30 Gy) actually induced expression of the three prime repair exonuclease 1 (Trex1) that degraded cytosolic DSDBs and attenuated the cGAS-STING pathway in tumor cells, thereby lowering production of IFN-β by tumor cells. In contrast, hypofractionated RT (8 Gy/day on three consecutive days) did not induce Trex1 and, therefore, promoted stronger cGAS-STING activity as well as higher expression of IFN-β in tumor cells. Interestingly, in this experimental system, knock-down of IFNAR1 in tumor cells completely abolished the therapeutic effects of this hypofractionated RT, indicating an indispensable role of tumor IFNAR1 expression [8]. Thus, the relative importance of IFN-I signaling in tumor cells may be determined by RT dose and fractionation. Nevertheless, host cell expression of IFNAR1 has been consistently shown to be instrumental for RT-induced immunomodulatory effects [9][8][10][11], encompassing increased infiltration of CD45+ hematopoietic cells (CD4+ and CD8+ T cells, DCs, and macrophages, etc.), upregulation of CXCL10 [11] and CXCL16 [12] (chemokines for CXCR3+ and CXCR6+ effector T cells, respectively), increase in Fas ligand (FasL), production of effector molecules GzmB and IFN-γ [11], upregulation of MHC molecules [13], and downregulation of don’t-eat-me signal CD47 [14] (the Yang effects of RT, Figure 1). Similar to host IFN-I signaling, host IFN-γ signaling has been shown to be essential in mediating anti-tumor immune responses elicited by RT using B16-OVA melanoma [15] and MC38 CRC [16]. While these studies compellingly pointed to a pivotal role of host IFN signaling in RT, it remains to be explored how the host JAK–STAT axis is involved in RT-induced immunological outcomes, although this is expected, given that JAKs and STATs have been shown to be crucial in dictating immune cell functions [17]. Further, a consensus on the role of tumor expression of IFNAR1 and IFNGR1 regarding RT response still cannot be reached, as either an essential role or a dispensable role was reported [6][8][7]. Additional work should be carried out to systematically assess how hyperfractionated RT, hypofractionated RT, and single-dose RT (high dose) may differentially impact the IFN-I and IFN-γ pathways in tumors, as distinct immunological outcomes may ensue after different RT regimens [6][8][7]. To this end, an early study reported that low-dose total-body irradiation (0.1–0.25 Gy, several times a week for a total dose of 1.5–2 Gy) induced impressive long-term tumor remission in the majority of patients with chronic lymphocytic leukemia and low-grade non-Hodgkin’s lymphoma. This was mediated by immune-enhancing mechanisms (e.g., T cell activation, increased production of IFN-γ and IL-2, and increased expression of IL-2R) rather than direct radiation killing [18]. Therefore, it may be possible that when RT dose is too high (>20 Gy), more immunosuppressive effects may be induced [8].
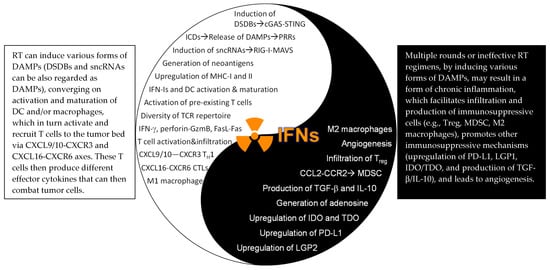
Figure 1. The Yin–Yang effects of the IFN–JAK–STAT in RT. DSDBs: double-strand DNA breaks; cGAS: cyclic GMP-AMP (cGAMP) synthase; STING: stimulator of interferon genes; ICDs: immunogenic cell death: DAMPs: damage-associated molecular patterns: sncRNAs: small endogenous non-coding RNAs; TH1: IFN-γ-producing type I CD4+ T cells; CTLs: cytotoxic T lymphocytes (CD8+ T cells); LGP2: laboratory of genetics and physiology 2; IDO/TDO: indoleamine/tryptophan 2,3-dioxygenase; MDSC: myeloid-derived suppressor cells. For further details, readers are encouraged to read our full-length review article, recently published in IJMS.
2. The IFN–JAK–STAT Axis in Radiosensitizing Effects of IOs.
It has long been known that immunological competence of patients contributes to the efficacy of RT, which determines the dose of radiation required for effective tumor control [19]. T cells, particularly CD8+ T cells, are required for therapeutic effects of RT, as demonstrated by a study wherein deletion of CD8+ T cells largely abolished RT-induced growth suppression of primary melanoma [20]. A subsequent study pinpointed that it was the pre-existing intratumoral T cells but not newly infiltrated T cells that governed the therapeutic effects of RT [21]. Since IOs can rejuvenate pre-existing T cells [22][23][24], it is expected that IOs could induce radiosensitizing effects and promote RT. In support of this idea, Rodriguez-Ruiz et al. showed that RT alone did not generate an overt abscopal effect (i.e., suppression of unirradiated tumors) in various tumor models (MC38 CRC, B16-OVA melanoma, and 4T1 triple-negative breast cancer), but adding ICBs to RT generated potent curative effects of both the irradiated and unirradiated tumors, achieving an impressive 100% cure of all the tumor-bearing mice [25]. Mechanistically, in vitro co-culture of activated CD8+ T cells with EMT-6 breast cancer cells greatly enhanced the radiosensitivity of EMT-6 cells, via IFN-γ production and iNOS upregulation [137]. Similarly, co-culture of CD4+ T cells with Hela cells radiosensitized Hela cells by promoting RT-induced G2/M arrest, in an IFN-γ-dependent fashion [138]. This IFN-γ-dependent mechanism was also confirmed in vivo. By promoting IFN-γ production by CD4+ and CD8+ T cells, immune checkpoint blockers (ICBs) (e.g., anti-CTLA-4, anti-PD-1/L1), arguably the most prominent development in the field of IOs over the past decade, significantly enhanced radiosensitivity [139,140]. IFN-γ, together with reduction of Treg after ICBs, promoted recruitment of tumor-associated eosinophils (TAEs), which then mediated normalization of tumor blood vessels [141]. This alleviated severe hypoxia in the TME, the most important mediator of radioresistance, and enhanced radiosensitivity. Convincingly, in vivo depletion of TAEs with anti-Siglec-F antibodies completely abrogated the therapeutic effect of anti-CTLA-4 [142]. Along this line, intratumoral injection of STING agonists also led to normalization of abnormal tumor vasculature, promoting infiltration of CD8+ T cells, in an IFN-I signaling-dependent fashion [143]. Interestingly, IFN-β itself was shown to possess anti-angiogenetic properties. By upregulating angiopoietin 1 (Angpt1), IFN-β suppressed abnormal angiogenesis and promoted tumor vascular maturation [144], which in turn enhanced intratumoral oxygenation and radiosensitivity [145]. While these studies collectively showed that blood vessel normalization represented an important mechanism of IO-induced radiosensitization, we reason that IOs may drive the radiosensitizing effects by metabolically reprogramming the TME. A recent study elegantly showed that ICBs tilted the intratumoral metabolic tug-of-war between TILs and tumor cells towards T cells [146] and suppressed the Warburg effect, a well-known metabolic feature of tumor cells that preferentially utilize glycolysis for their bioenergetic and biosynthetic needs. The suppression of the Warburg effect in tumor cells reduced accumulation of lactate, pyruvate, and other antioxidants such as glutathione, thereby facilitating ROS production and oxidative stress induced by RT. Greater intracellular ROS level would help fix oxidative damage to DNA and enhance radiosensitivity. In support of this idea, knock-down of STAT1 predisposed nu61 HNSCC cells to RT-driven suppression of glycolysis and reduction in anti-oxidation capacity, leading to greater tumor growth suppression and radiosensitization [67], but it remains to be tested how IOs affect STAT1 and other members of the JAK-STAT family in tumor cells. In addition, IOs induced production of effector molecules in addition to IFNs such as TNF-α, perforin, and GzmB, and engaged other cell death pathways (e.g., FasL-Fas), which may facilitate or amplify the direct and indirect effects of RT, resulting in radiosensitization.
3. The IFN–JAK–STAT Axis Underscoring Combination Therapy of RT and IOs
With the immunogenic effects from RT and the radiosensitizing consequences from IOs, it is an appealing idea to combine RT and IOs as a strategy to improve therapeutic efficacy. To this end, adding anti-PD-L1 to RT significantly boosted overall efficacy and enhanced anti-tumor immunity, including increased production of IFN-γ in pancreatic ductal adenocarcinoma [26] and hepatocellular carcinoma [27]. Similar results were also found in TUBO mammary and MC38 CRC tumors when treated with anti-PD-L1 and RT, which greatly promoted CTL activation and production of IFN-γ and TNF-α. This, in turn, reduced infiltration of immunosuppressive MDSCs, primarily mediated by TNF-α and, to a minor extent, by IFN-γ [7]. Another study tested combination therapy of anti-CTLA-4 with RT and demonstrated that this strategy not only depleted Treg (by anti-CTLA-4) but also promoted the diversity of TCR repertoire of TILs (by RT), resulting in potent anti-tumor responses. However, upregulation of PD-L1 on tumor cells rendered the majority of tumor-bearing mice resistant to RT+anti-CTLA-4, and additional blocking of PD-1/PD-L1 (triple-therapy) significantly improved the overall efficacy [28]. In a follow-up study, this same group reported a PD-L1-independent resistance mechanism that was regulated by STAT1-mediated epigenetic changes, which, when targeted with an inhibitor (Ruxolitinib), successfully overcame the therapeutic resistance [29]. Clinically, greater benefits have been reported in various tumor types when treated with combination therapies of RT+IOs, providing proof-of-concept clinical evidence [30]. From a mechanistic standpoint, although pre-existing intratumoral T cells play a predominant role in RT alone [21], both pre-existing and infiltrated T cells were required for the combination therapy of RT+IOs [31]. In keeping with an essential role of host IFNAR1 signaling in mediating RT and IOs, either use of IFNAR1KO mice [8] or in vivo blocking of IFNAR1 with neutralizing antibodies [10] completely abolished the synergistic effects of RT+IOs. On the other hand, although T cells and host IFN-γ signaling are essential for RT [15] and ICBs [22], to the best of our knowledge, their importance in combination therapy of RT and IOs has not been established. Likewise, very limited efforts, if any, have been made to evaluate the role of the JAK-STAT in RT+IOs. Therefore, despite our improved mechanistic understanding of the role of IFN–JAK–STAT axis in RT+IOs, more work needs to be carried out to pinpoint their involvement in different experimental settings. To this end, another study showed that activation of STING after a single-dose 20 Gy RT in the MC38 CRC model actually drove an influx of immunosuppressive MDSCs and Treg, in an IFN-I and CCR2-dependent manner [32]. Moreover, RT, especially multiple rounds or prolonged regimens, can induce adaptive immunosuppression such as upregulation of PD-L1 and CD47 by promoting sustained IFN signaling in tumor cells [13] and production of immunosuppressive cytokine TGF-β [33] (the Yin effects of RT, Figure 1). Thus, rational strategies are needed to combine RT with IOs, by maximizing immunostimulatory effects and concomitantly minimizing immunosuppressive effects of RT, which will greatly improve therapeutic efficacy. We argue that the final outcomes may well depend on the specific tumor types and stages, radiation dose, fractionation, and types of RT (particles vs. photon), and the scheduling of RT and IOs.
References
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842.
- Ranoa, D.R.; Parekh, A.D.; Pitroda, S.P.; Huang, X.; Darga, T.; Wong, A.C.; Huang, L.; Andrade, J.; Staley, J.P.; Satoh, T.; et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 2016, 7, 26496–26515.
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059.
- Xu, P.; Xu, Y.; Hu, B.; Wang, J.; Pan, R.; Murugan, M.; Wu, L.J.; Tang, Y. Extracellular ATP enhances radiation-induced brain injury through microglial activation and paracrine signaling via P2X7 receptor. Brain Behav. Immun. 2015, 50, 87–100.
- Wilkins, A.C.; Patin, E.C.; Harrington, K.J.; Melcher, A.A. The immunological consequences of radiation-induced DNA damage. J. Pathol. 2019, 247, 606–614.
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496.
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852.
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology 2017, 6, e1339857.
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003.
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017, 77, 839–850.
- Lim, J.Y.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol. Immunother. 2014, 63, 259–271.
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107.
- Garnett, C.T.; Palena, C.; Chakraborty, M.; Tsang, K.Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994.
- Vermeer, D.W.; Spanos, W.C.; Vermeer, P.D.; Bruns, A.M.; Lee, K.M.; Lee, J.H. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int. J. Cancer 2013, 133, 120–129.
- Lugade, A.A.; Sorensen, E.W.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J. Immunol. 2008, 180, 3132–3139.
- Gerber, S.A.; Sedlacek, A.L.; Cron, K.R.; Murphy, S.P.; Frelinger, J.G.; Lord, E.M. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol. 2013, 182, 2345–2354.
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384.
- Safwat, A. The immunobiology of low-dose total-body irradiation: More questions than answers. Radiat. Res. 2000, 153, 599–604.
- Makidono, R. Decrease in radio-sensitivity of the tumor by radiation-induced damage to immuno-related cells. Gan No Rinsho Jpn. J. Cancer Clin. 1987, 33, 1229–1237.
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595.
- Arina, A.; Beckett, M.; Fernandez, C.; Zheng, W.; Pitroda, S.; Chmura, S.J.; Luke, J.J.; Forde, M.; Hou, Y.; Burnette, B.; et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun. 2019, 10, 3959.
- Shi, L.Z.; Fu, T.; Guan, B.; Chen, J.; Blando, J.M.; Allison, J.P.; Xiong, L.; Subudhi, S.K.; Gao, J.; Sharma, P. Interdependent IL-7 and IFN-gamma signalling in T-cell controls tumour eradication by combined alpha-CTLA-4+alpha-PD-1 therapy. Nat. Commun. 2016, 7, 12335.
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280.
- Peggs, K.S.; Quezada, S.A.; Korman, A.J.; Allison, J.P. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 2006, 18, 206–213.
- Rodriguez-Ruiz, M.E.; Rodriguez, I.; Garasa, S.; Barbes, B.; Solorzano, J.L.; Perez-Gracia, J.L.; Labiano, S.; Sanmamed, M.F.; Azpilikueta, A.; Bolaños, E.; et al. Abscopal Effects of Radiotherapy Are Enhanced by Combined Immunostimulatory mAbs and Are Dependent on CD8 T Cells and Crosspriming. Cancer Res. 2016, 76, 5994–6005.
- De Ridder, M.; Jiang, H.; Van Esch, G.; Law, K.; Monsaert, C.; Van den Berge, D.L.; Verellen, D.; Verovski, V.N.; Storme, G.A. IFN-gamma+ CD8+ T lymphocytes: Possible link between immune and radiation responses in tumor-relevant hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 647–651.
- Wang, Y.; Radfar, S.; Khong, H.T. Activated CD4+ T cells enhance radiation effect through the cooperation of interferon-gamma and TNF-alpha. BMC Cancer 2010, 10, 60.
- Zheng, X.; Fang, Z.; Liu, X.; Deng, S.; Zhou, P.; Wang, X.; Zhang, C.; Yin, R.; Hu, H.; Chen, X.; et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Invest. 2018, 128, 2104–2115.
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254.
- Carretero, R.; Sektioglu, I.M.; Garbi, N.; Salgado, O.C.; Beckhove, P.; Hämmerling, G.J. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat. Immunol. 2015, 16, 609–617.
- Zheng, X.; Zhang, N.; Qian, L.; Wang, X.; Fan, P.; Kuai, J.; Lin, S.; Liu, C.; Jiang, W.; Qin, S.; et al. CTLA4 blockade promotes vessel normalization in breast tumors via the accumulation of eosinophils. Int. J. Cancer 2020, 146, 1730–1740.
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest. 2019, 129, 4350–4364.
- Dickson, P.V.; Hamner, J.B.; Streck, C.J.; Ng, C.Y.; McCarville, M.B.; Calabrese, C.; Gilbertson, R.J.; Stewart, C.F.; Wilson, C.M.; Gaber, M.W.; et al. Continuous delivery of IFN-beta promotes sustained maturation of intratumoral vasculature. Mol. Cancer Res. 2007, 5, 531–542.
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340.
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241.
- Pitroda, S.P.; Wakim, B.T.; Sood, R.F.; Beveridge, M.G.; Beckett, M.A.; MacDermed, D.M.; Weichselbaum, R.R.; Khodarev, N.N. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009, 7, 68.
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180.
- Kim, K.J.; Kim, J.H.; Lee, S.J.; Lee, E.J.; Shin, E.C.; Seong, J. Radiation improves antitumor effect of immune checkpoint inhibitor in murine hepatocellular carcinoma model. Oncotarget 2017, 8, 41242–41255.
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12.
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185.
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.L.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res. 2017, 23, 5514–5526.
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736.
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242.
More
Information
Subjects:
Immunology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
759
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No