 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raquel Iglesias-Fernández | + 1954 word(s) | 1954 | 2020-06-10 10:15:45 | | | |
| 2 | Catherine Yang | -8 word(s) | 1946 | 2020-10-27 09:34:48 | | |
Video Upload Options
The seed is the propagule of higher plants and allows its dissemination and the survival of the species. Seed dormancy prevents premature germination under favourable conditions. Dormant seeds are only able to germinate in a narrow range of conditions. During after-ripening (AR), a mechanism of dormancy release, seeds gradually lose dormancy through a period of dry storage. Part of this review focuses on how the methylation of mRNA and genomic DNA affects gene expression upon late stages of seed development, especially during dormancy. DNA methylation is a well-known epigenetic mechanism of controlling gene expression. In Arabidopsis thaliana, while there is a global increase in CHH-context methylation through embryogenesis, global DNA methylation levels remain stable during seed dormancy, decreasing when germination occurs. The biological significance of nucleic acid methylation upon seed development is discussed.
1. The Seed Epitranscriptome
Non-classical chemical changes, apart from capping and polyadenylation modifications, alter the stability of mRNAs,. More than 160 chemical modifications of mRNAs have been recently reported in A. thaliana, as a whole they are considered the epitranscriptome [1]. These alterations include the methylation of the adenosine and cytosine nucleotides: N6-methyladenosine (m6A) and 5-methylcytosine (m5C) [2]. These mRNA modifications modulate transcript stability, and they have been found in transcripts that are being degraded, suggesting they are involved in mRNA turnover. However, stable mRNAs contain chemical modifications related to the alternative splicing of introns. This fact indicates that these modifications could also regulate the expression of different transcript variants [3]. mRNA chemical modification has been recently described as essential for plant development, and it is considered a new emerging level of controlling gene expression [1]. In A. thaliana, the first mRNA methylation map has revealed the presence of thousands of methylated mRNAs [4]. A new molecular system formed by RBPs has been involved in adding (writers), removing (erasers) and recognizing (readers) chemical modifications present at the mRNA transcripts. The A. thaliana RNA Adenosine Methylase MTA (writer) has been described to be essential for plant survival as null mta mutants are embryo-lethal [5]. MTA is mainly expressed in dividing tissues, such as seeds upon development. Moreover, the expression of MTA under the control of the seed-specific ABI3 promoter rescues the lethality of null mta mutants, indicating its relevance during seed development [6].
2. Regulation of Seed Development through DNA Methylation
2.1. DNA Methylation Basis
The chemical modification of DNA by the addition of a methyl group at the fifth position on the pyrimidine ring of cytosines is a stable and reversible epigenetic mark [7]. Centromeres and repeated sequences are heterochromatic regions, characterized by heavily methylated DNA [8]. DNA methylation in euchromatic regions and within genes has different effects on gene expression. While methylation within transcribed regions (the body of genes, mostly methyl-CG) is associated with constitutive expression levels [9], DNA methylation within promoters is associated with repression, i.e., gene silencing [10] and tissue-specific expression [8]. DNA methylation is a conserved epigenetic mechanism that, in plants, occurs at three different sequence contexts: the symmetrical CG and CHG, and the asymmetrical CHH, where H can be a cytosine, a thymidine or an adenine. Symmetrical methylation can be maintained during the cell cycle by DNA METHYLTRANSFERASE 1 (MET1) and CHROMOMETHYLASE 3 (CMT3), which catalyse DNA methylation on hemi-methylated DNA strands [11]. Asymmetrical methylation requires re-establishment after each DNA replication round by persistent de novo methylation by DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) at transposons and other repeated sequences in euchromatic chromosome arms. In this case, small RNAs cause transcriptional gene silencing by directing the addition of DNA methylation to specific DNA sequences by nucleotide homology mediated by ARGONAUTE (AGO) proteins, in a pathway called RNA-directed DNA methylation (RdDM) [12]. CHROMOMETHYLASE 2 (CMT2) catalyses CHH methylation at histone H1-containing heterochromatin, where DRM2 is inhibited [13]. Alternatively, sequences that have been methylated can become demethylated either passively or actively. Passive demethylation is due to subsequent rounds of DNA replication without the maintenance of the methylation pattern. Active demethylation is catalysed by DNA glycosylases —DEMETER (DME), REPRESSOR OF SILENCING1/DEMETER-LIKE 1 (ROS1), DEMETER-LIKE 2 (DML2) and DEMETER-LIKE 3 (DML3), in A. thaliana [14].
2.2. DNA-Methylation and Seed Development
The initial observations of cell nuclei, during A. thaliana seed maturation, have shown small size and very condensed chromatin, while the contrary happens during seed germination, suggesting a change in the balance between heterochromatin and euchromatin in the transition to germination [15]. Recent whole-genome bisulfite sequencing analysis supports these previous observations and shows that DNA methylation is dynamic during the process of seed development [16][17][18][19][20] (Figure 1). DNA methylation is known to play a critical role in seed development since mutations in MET1 and DME have been described to affect this developmental process [21][22]. Now, we know that during seed development there is a specific increase in global non-CG methylation, mainly due to changes in methyl-CHH and especially at transposable elements (TEs), both in Glycine max and A. thaliana [16][17][18][19][20]. This increase in global non-CG methylation occurs simultaneously throughout the seed, while CG and CHG methylations do not change significantly during seed development or in any specific part of it [16][17][19][20]. In mature embryos, CHH hypermethylation in TEs matches DNA hypomethylation in the endosperm. A role for RdDM in the establishment of this methylation is supported by the fact that 24-nt siRNAs derived from these regions are most abundant during late embryogenesis, coinciding with elevated expression of RdDM components [18]. Analysis of drm1 drm2 cmt3 (ddc) and drm1 drm2 cmt3 cmt2 (ddcc) mutants shows that CMT2 is also involved in this TE hypermethylation by the identification of over 100 de-repressed TEs which show different CHH methylation compared to that of randomly selected TEs [16]. This fact reinforces the idea that non-CG methylation and, specifically, methyl-CHH, may be an important mechanism to avoid TE activity during seed maturation [19]. Regarding DNA methylation within genes, it has been found that those genes that are transcribed are more likely to be differentially methylated during seed maturation than non-expressed genes [17]. The majority of these differentially methylated genes become transcriptionally down-regulated as seed maturation proceeds—DNA methylation levels increase—pointing to a specific role of DNA methylation in the repression of specific genes [17].
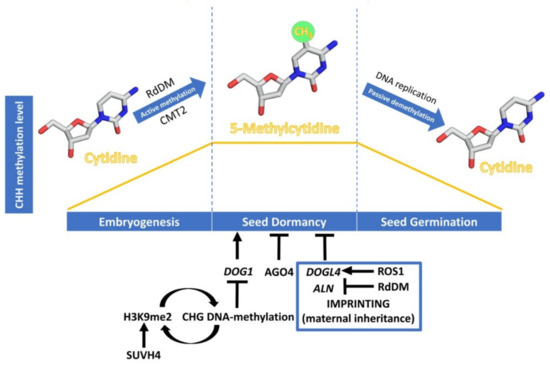
Figure 1. Dynamics of CHH DNA methylation (orange line) during seed development, and epigenetic regulation of seed dormancy. The DNA methylation process has been schematised with cytidine and 5-methylcytidine molecules designed using Chimera 1.14 [23]. Epigenetic regulation of seed dormancy has been schematised with arrows indicating a promotive effect and bars indicating a repressive effect. Regulation through genomic imprinting is indicated within the blue box.
The expression levels of DOG1 are regulated by various molecular mechanisms like alternative splicing, selective polyadenylation, and non-coding RNAs [24][25], but in this review we will highlight the consequences of DOG1 H3K9 and DNA methylation in the regulation of seed dormancy. DNA methylation in the CHG context is perpetuated in A. thaliana by the SUVH family of histone H3 lysine 9 N-methyltransferases. Their SRA domain binds to methylated CHG cytosines, leading to H3K9me2 deposition. Conversely, sites of H3K9 methylation recruit DNA methyltransferases CMT3 and CMT2, thus forming a self-reinforcing loop of repressive epigenetic marks. The seeds of SUVH4 mutants (also known as kyp) show enhanced dormancy and up-regulation of DOG1 and ABI3 [26], supporting that histone H3K9me2 caused by KYP/SUVH4 induces their silencing through DNA methylation, negatively affecting seed dormancy. A possible role of the RdDM pathway in the silencing of seed dormancy genes has been suggested from studies in cereals. In barley, the AGO4_9 gene AGO1003 is expressed differentially in the embryos of dormant and non-dormant grains [27]. In wheat, AGO802B, a wheat ortholog of an AGO4_9, is expressed during grain development and its expression is significantly lower in preharvest sprouting-resistant varieties than in susceptible ones; implying that DNA methylation levels could be reduced in PHS susceptible varieties compared to resistant lines [28]. This result indicates that AGO4 is a negative regulator of dormancy, although it is not known whether specific coding genes are subjected to silencing through RdDM or which ones are. Recently, it has been demonstrated that mutants with loss or reduced function of the chromatin-remodelling factor PICKLE (PKL) show increased seed dormancy; PKL directly represses DOG1 expression [29].
Genomic imprinting is an epigenetic mechanism that alters gene expression in a parent-of-origin manner. As a consequence, the expression of some genes depends on whether the allele was inherited from the maternal or the paternal parent [30]. Reciprocal crosses between A. thaliana accessions with increased seed dormancy—Cvi—and with reduced seed dormancy—C24—have suggested a maternal inheritance of seed dormancy levels. Thus, the two populations of F-1 hybrid seeds from these reciprocal crosses exhibit distinct levels of dormancy, which tend to phenocopy the maternal traits. In parallel, 57 dormancy-specific maternally expressed genes have been analysed [31], but the molecular mechanisms underpinning the regulation of these genes are yet to be elucidated. Recent studies have shown the importance of genomic imprinting, through DNA-methylation, in regulating the expression in the endosperm of two maternally expressed negative regulators of seed dormancy, DOG1-LIKE 4 (DOGL4) and ALLANTOINASE (ALN) [32][33]. In A. thaliana, active DNA demethylation depends on the activity of ROS1, which directly excises methyl-C from DNA [14]. Interestingly, ros1 mutants are hypersensitive to ABA during early seedling development [34], showing that ROS1-driven DNA demethylation regulates seed dormancy, and the response to ABA by controlling the expression of DOGL4, a negative regulator of seed dormancy and the ABA response. ROS1 appears to antagonize the RdDM pathway, required for the imprinting that underlies the suppression of the paternal allele, by reducing the high level of DNA methylation of the paternal DOGL4 promoter, and consequently the gene repression. While both dogl4 and ros1 mutants show a decrease in germination rates and an increase in the response to ABA, overexpression of DOGL4 in ros1 mutant background reverts the enhanced seed dormancy and ABA hypersensitivity phenotype, which shows that ROS1 regulation of seed dormancy is via DOGL4 [32].
DNA methylation also plays an important role in regulating the preferential maternal ALN expression in the seed endosperm. Higher CHH methylation levels in a transposable element present in the 5′ region of ALN, targeted by RdDM, correlate with lower expression of the gene. Paternal allele DNA methylation appears to be established during the gametogenesis in sperm cells in this gene [33]. During seed development, cold induces further CHH methylation in this region. This mechanism might allow seeds to store information about cold temperatures and optimize the germination timing. Upon germination, cold-induced DNA methylation is lost, allowing optimal gene expression in the next generation [107]. Further studies are required to understand how the RdDM machinery can discriminate paternal from maternal alleles and maintain the different levels of methylation after fertilization. It is suggested that the RdDM machinery might act preferentially at methylated DNA or that secondary imprinting such as histone modification might direct RdDM to the paternal allele [33].
During germination, although CG and CHG methylations remain stable, which indicates that MET1 and CMT3 are active in maintenance, there is a drop in CHH methylation levels. There is, however, no decrease in siRNAs for these hypomethylated loci nor in RdDM machinery components abundance, which points to demethylation causing the drop [16][20]. DNA demethylases are weakly or not expressed during germination, and demethylases mutants show no significant changes in DNA methylation when compared to wild type [16]. This suggests that passive demethylation may be behind the decrease in DNA methylation levels during the process of germination, although further studies are still needed to support this assertion. More than 12,000 differential methylated regions have been found in analyses upon A. thaliana seed germination, with a different degree of methylation in promoters of over 1800 differently expressed genes [20]. This implies that the dynamics of DNA methylation can regulate germination-related gene expression. Nevertheless, further studies on the specific set of genes regulated by changes in DNA methylation levels are required to understand the importance of this epigenetic regulation.
References
- Shen, L.; Liang, Z.; Wong, C.E.; Yu, H. Messenger RNA modifications in plants. Trends Plant Sci. 2019, 24, 328–341.
- Cui, X.; Liang, Z.; Shen, L.; Zhang, Q.; Bao, S.; Geng, Y.; Zhang, B.; Leo, V.; Vardy, L.A.; Lu, T.; et al. 5-Methylcytosine RNA methylation in Arabidopsis thaliana. Mol. Plant 2017, 10, 1387–1399.
- Vandivier, L.E.; Campos, R.; Kuksa, P.P.; Silverman, I.M.; Wang, L.-S.; Gregory, B.D. Chemical modifications mark alternatively spliced and uncapped messenger RNAs in Arabidopsis. Plant Cell 2015, 27, 3024–3037.
- Fray, R.G.; Simpson, G.G. The Arabidopsis epitranscriptome. Curr. Opin. Plant Biol. 2015, 27, 17–21.
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA Iis an Arabidopsis Messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288.
- Bodi, Z.; Zhong, S.; Mehra, S.; Song, J.; Graham, N.; Li, H.; May, S.; Fray, R.G. Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front. Plant Sci. 2012, 3.
- Diez, C.M.; Roessler, K.; Gaut, B.S. Epigenetics and plant genome evolution. Curr. Opin. Plant Biol. 2014, 18, 1–8.
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201.
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110.
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69.
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220.
- Matzke, M.A.; Kanno, T.; Matzke, A.J.M. RNA-Directed DNA methylation: The evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 2015, 66, 243–267.
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506.
- Parrilla-Doblas, J.T.; Roldán-Arjona, T.; Ariza, R.R.; Córdoba-Cañero, D. Active DNA Demethylation in plants. Int. J. Mol. Sci. 2019, 20, 4683.
- van Zanten, M.; Koini, M.A.; Geyer, R.; Liu, Y.; Brambilla, V.; Bartels, D.; Koornneef, M.; Fransz, P.; Soppe, W.J.J. Seed maturation in Arabidopsis thaliana is characterized by nuclear size reduction and increased chromatin condensation. Proc. Natl. Acad. Sci. USA 2011, 108, 20219–20224.
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171.
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 2017, 7.
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179.
- Lin, J.-Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.-F.; Chen, P.-Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739.
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Lister, R.; Lewsey, M.G.; Whelan, J. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017, 18.
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42.
- Xiao, W.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Regulation of seed size by hypomethylation of maternal and paternal genomes. Plant Physiol. 2006, 142, 1160–1168.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612.
- Carrillo-Barral, N.; del Carmen Rodríguez-Gacio, M.; Matilla, A.J. Delay of Germination-1 (DOG1): A key to understanding seed dormancy. Plants 2020, 9, 480.
- Li, P.; Ni, H.; Ying, S.; Wei, J.; Hu, X. Teaching an old dog a new trick: Multifaceted strategies to control primary seed germination by DELAY OF GERMINATION 1 (DOG1). Phyton (B. Aires). 2020, 89, 1–12.
- Zheng, J.; Chen, F.; Wang, Z.; Cao, H.; Li, X.; Deng, X.; Soppe, W.J.J.; Li, Y.; Liu, Y. A novel role for histone methyltransferase KYP/SUVH4 in the control of Arabidopsis primary seed dormancy. New Phytol. 2012, 193, 605–616.
- Singh, M.; Singh, J. Seed development-related expression of ARGONAUTE4_9 class of genes in barley: Possible role in seed dormancy. Euphytica 2012, 188, 123–129.
- Singh, M.; Singh, S.; Randhawa, H.; Singh, J. Polymorphic homoeolog of key gene of RdDM pathway, ARGONAUTE4_9 class is associated with pre-harvest sprouting in Wheat (Triticum aestivum L.). PLoS ONE 2013, 8.
- Zha, P.; Liu, S.; Li, Y.; Ma, T.; Yang, L.; Jing, Y.; Lin, R. The evening complex and the chromatin-remodeling factor PICKLE coordinately control seed dormancy by directly repressing DOG1 in Arabidopsis. Plant Commun. 2020, 1, 100011.
- Köhler, C.; Wolff, P.; Spillane, C. Epigenetic mechanisms underlying genomic imprinting in plants. Annu. Rev. Plant Biol. 2012, 63, 331–352.
- Piskurewicz, U.; Iwasaki, M.; Susaki, D.; Megies, C.; Kinoshita, T.; Lopez-Molina, L. Dormancy-specific imprinting underlies maternal inheritance of seed dormancy in Arabidopsis thaliana. eLife 2016.
- Zhu, H.; Xie, W.; Xu, D.; Miki, D.; Tang, K.; Huang, C.-F.; Zhu, J.-K. DNA demethylase ROS1 negatively regulates the imprinting of DOGL4 and seed dormancy in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2018, 115, E9962–E9970.
- Iwasaki, M.; Hyvärinen, L.; Piskurewicz, U.; Lopez-Molina, L. Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy. eLife 2019, 8.
- Kim, J.-S.; Lim, J.Y.; Shin, H.; Kim, B.-G.; Yoo, S.-D.; Kim, W.T.; Huh, J.H. ROS1-dependent DNA demethylation is required for ABA-inducible NIC3 expression. Plant Physiol. 2019, 179, 1810–1821.


