Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gaetano Aurilio | + 1267 word(s) | 1267 | 2021-10-26 07:51:15 | | | |
| 2 | Rita Xu | Meta information modification | 1267 | 2021-11-05 02:32:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Aurilio, G. Renal Cell Carcinoma Metabolism. Encyclopedia. Available online: https://encyclopedia.pub/entry/15719 (accessed on 07 February 2026).
Aurilio G. Renal Cell Carcinoma Metabolism. Encyclopedia. Available at: https://encyclopedia.pub/entry/15719. Accessed February 07, 2026.
Aurilio, Gaetano. "Renal Cell Carcinoma Metabolism" Encyclopedia, https://encyclopedia.pub/entry/15719 (accessed February 07, 2026).
Aurilio, G. (2021, November 04). Renal Cell Carcinoma Metabolism. In Encyclopedia. https://encyclopedia.pub/entry/15719
Aurilio, Gaetano. "Renal Cell Carcinoma Metabolism." Encyclopedia. Web. 04 November, 2021.
Copy Citation
Renal cell carcinoma (RCC) is highly dependent on dysregulated expression of several genes, and the greater understanding of metabolomic RCC profiling has played a key role in optimizing the therapeutic management of patients in clinical practice.
renal cell carcinoma
metabolomic profiling
therapeutics
1. Background
Renal cell carcinoma (RCC) is highly dependent on dysregulated expression of several genes, and the greater understanding of metabolomic RCC profiling has played a key role in optimizing the therapeutic management of patients in clinical practice. Histopathological classification of RCC distinguishes three most frequent subtypes: clear cell RCC (ccRCC), which accounts for the 70–75% of all RCC; papillary RCC (10–15%); and chromophobe RCC (5%) [1]. The phosphatidylinositol-3 kinase (PI3K)/protein kinase B (Akt) signaling has been considered as notorious in the main critical pathway for carcinogenesis in RCC, especially for ccRCC histology. The serine/threonine kinase Akt is the most relevant downstream member of this pathway, having a crucial transduction role throughout the signaling cascade [2]. Until now, it was estimated that at least 17 genes were strictly correlated with RCC development, and alterations of these genes have been suggested to elicit a dysregulation of various cellular metabolisms involved in oxygen, energy, and nutrient control. Data on cancer cell metabolism have shown that cancer cells are prone to a metabolic energy reprogramming for supporting cell growth and division, and such metabolism reprogramming has been listed as one of the cornerstones of cancer development [3]. Along this line, the Warburg effect—otherwise known as aerobic glycolysis—is one of the first metabolic alterations to be detected, and is present in the majority of cancer cells. It displays the attitude of tumor cells to prefer glycolysis in place of oxidative phosphorylation (OXPHOS), even when oxygen is available [4], a process maximally expressed in ccRCC [5]. Epigenetic mechanisms have also been recognized for deregulating certain metabolic processes implicated in growth and cell survival of RCC [6].
2. Outline of Renal Cell Carcinoma Metabolism
2.1. Metabolic Reprogramming
Glucose metabolism is of key importance in cancer development. Glycolysis is a metabolic pathway that converts glucose into pyruvate, finally generating energy to sustain cellular growth. Under normal oxygen condition, glucose is metabolized in pyruvate, and the latter produces the molecule acetyl-coenzyme A, which fuels the OXPHOS pathway. The entire process culminates into the synthesis of adenosine triphosphate (ATP), the most energetic compound for almost all metabolic reactions. In this scenario of normoxia, lactate production is minimal and basal rate of glycolysis occurs; conversely, under low oxygen conditions, glycolysis is facilitated, and pyruvate is converted into lactate [7].
Cancer cells benefit from different oxygen levels due to a well-known chaotic tumor vasculature. However, regardless of this, cancer cells display a highly aerobic glucose consumption, a key feature of cancer metabolism known as aerobic glycolysis, or the Warburg effect. Data from Warburg and coworkers demonstrated that cancer cells required more glucose than healthy cells, in a ratio of approximately 10 to 1, and originated a greater production of lactate [4]. This occurred through both increased glycolysis and with a sufficient oxygen supply.
In ccRCC, the upregulation of the PI3K/Akt pathway is the major cause of the Warburg effect. The autophosphorylation of PI3K induces Akt activation through PIP2-to-PIP3 conversion: the Akt-mediated signaling cascade leads to phosphorylation of target downstream effectors (genes and proteins) able to promote the Warburg effect, and therefore a wide range of cellular functions, as illustrated in Figure 1. Akt activation induces stimulation of Bcl-2 and inhibition of Bax, helping cancer cell survival and accordingly their resistance [8]. Akt overexpression also inactivates certain FOX-family members that play a role in mechanisms of drug resistance. In addition, Akt is able to overactivate the NF-kB system, which in turn fuels the growth of cancer cells, thus promoting drug resistance [9]. Akt signaling activates the hypoxia-inducible transcription factors (HIF) family, thus increasing the transcription of glucose transporters, such as GLUT, and of most glycolytic enzymes, such as PDK and PKM2, finally promoting aerobic glycolytic metabolism, and leading to increased lactate production and OXPHOS inactivity.
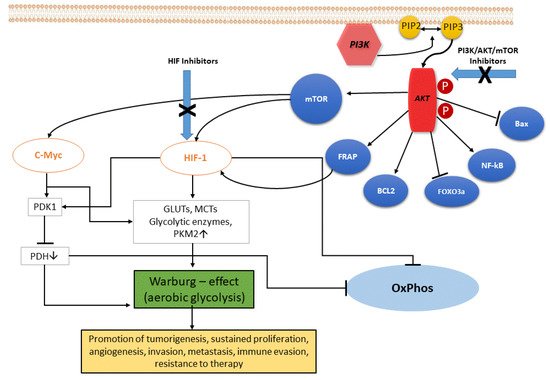
Figure 1. The PI3K/Akt signaling pathway. PI3K, phosphatidylinositol 3 kinase; Akt, protein kinase HIF, hypoxia-induced factor; FoxO3a, Forkhead box O3; mTOR, mammalian target of rapamycin; FRAP, FKBP-rapamycin associated protein; BCL2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; eNOS, endothelial cell nitric oxide synthase; cMyc, cellular Myc oncogene; HIF, hypoxia-inducible factor; GLUTs, glucose transporters; MCTs, monocarboxylate transporters; PKM2, pyruvate kinase muscle isozyme M2; PDH, pyruvate dehydrogenase (complex); PDK1, pyruvate dehydrogenase kinase 1; OxPhos, oxidative phosphorylation.
As a consequence, some advantages for cancer cells do occur. Firstly, lipid synthesis, as well as amino acid and nucleotide production, are sustained through the glycolysis process, which supports cell growth in a condition of oxygen lability. Additionally, the Warburg phenomenon decreases reactive oxygen species (ROS) levels by inducing antioxidant glutathione, thus helping cells against oxidative stress. Lastly, increased lactate production via glycolysis generates an acidic tumor microenvironment, which has been correlated with aggressive cancer phenotype, specifically a high risk for recurrences, as well as occurrence of metastases [10].
In the tumor microenvironment, lactate induces HIF-1-alpha stabilization, and hence cancer and endothelial cells respectively exhibit hyperexpression of vascular endothelial growth factor (VEGF) and its type-2 receptor (VEGFR2) [11]. Accordingly, cancer invasion, migration, angiogenesis, and overall growth and cell proliferation are sustained. Of interest, high lactate expression in tumor microenvironment reduces the activity of T lymphocytes and natural killer cells, as well as cytokine release by dendritic cells, overall displaying an immunosuppressive action [12]. Therefore, lactate was thus configured as a metabolic cancer enhancer. Hence, the angiogenic and glycolytic phenotypes in ccRCC are strictly connected to one another, and often coexist in the same tumor. RCC has been defined as a “metabolic disease” for its characteristic metabolic defects and alterations that are consequences of the unique genetic background that drive this cancer [13].
2.2. Epigenetic Alterations
Epigenetic alterations are chromatin impairments that cause reversible modifications in gene expression sequences. In normal tissue, epigenetic mechanisms are able to provide genomic balance, as well as chromosome regulation.
Contrarily, along with several other factors, metabolic boosts can induce abnormal epigenetic changes that may contribute to triggering a malignant transformation. An altered epigenetic regulation is a distinctive trait of cancer growth, and encompasses both proto-oncogene activation and onco-suppressor gene downregulation. It can be sustained by certain mechanisms, such as DNA methylation, histone modifications, and chromatin remodeling, briefly illustrated here as follows. The first one is a key, well-investigated epigenetic mechanism. Cancer cells exhibit extensively hypomethylated DNA, which is responsible for genomic instability, the latter characterized by high mutational burden, proto-oncogene induction, and loss of genomic imprinting. An abnormal hypermethylated status instead occurs for gene promoters, and correlates with reduced transcription expression of onco-suppressor genes. The second one deals with post-translational changes of histone tails, mainly as methylation and acetylation of specific amino acid sequences. These processes are regulated by modification-inducing enzymes called histone methyltransferases and histone acetyltransferases, respectively, and by enzymes removing such modifications, also called lisine demethylases and histone deacetylases (HDAC), respectively. The third one regards chromatin remodeling complexes, particularly through inactivating mutations occurring in polybromo-1 (PBRM1) and BRCA-associated protein-1 (BAP1) genes. PBRM1 is a chromatin regulating gene that modulates the accessibility to DNA transcription, and data support PBRM1 inactivation as a promoter for cell proliferation and migration. BAP1 is a tumor suppressor gene and coregulates many biological mechanisms, from cell cycle function to cell death, passing through metabolic processes and DNA regulation. RCC with BAP1 deficiency is significantly associated with decreased survival [14]. Therefore, taking into account all of the above, abnormal DNA methylation, histone post-translational changes, and chromatin remodeling complexes are the epigenetic signature of RCC, prone to control transcription and DNA integrity via chromatin modifications, and thus culminating in increased carcinogenesis and tumor aggressiveness.
References
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105.
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Warburg, O. The metabolism of carcinoma cells. Cancer Res. 1925, 9, 148–163.
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419.
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49.
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157.
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36.
- Kumar, S.; Kushwaha, P.P.; Gupta, S. Emerging targets in cancer drug resistance. Cancer Drug Resist. 2019, 2, 161–177.
- Lameirinhas, A.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C. The Complex Interplay between Metabolic Reprogramming and Epigenetic Alterations in Renal Cell Carcinoma. Genes 2019, 10, 264.
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418.
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52.
- Rathmell, W.K.; Rathmell, J.C.; Linehan, W.M. Metabolic Pathways in Kidney Cancer: Current Therapies and Future Directions. J. Clin. Oncol. 2018.
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
700
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No