 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Timothy Hurst Pohlman | + 1705 word(s) | 1705 | 2021-10-27 06:28:34 | | | |
| 2 | Jason Zhu | Meta information modification | 1705 | 2021-11-03 02:22:24 | | |
Video Upload Options
The pathophysiology of hemorrhagic shock involves a decrease in systemic oxygen delivery to a level less than what is required to maintain cellular function.
1. Introduction
For the polytrauma patient, brain injury is the most common cause of early death followed by acute blood loss as the second most common cause of early death [1][2]. In the U.S., 150,000 people die each year due to injury and many of these deaths occur in relatively younger individuals, which causes an aggregate loss of productive life of over 3.3 million years [3]. This results in an annual cost to society of USD 2.34 billion in today’s dollars from lost wages and medical costs. In prospective studies that examine resuscitation after trauma the median time to hemorrhagic death is 2.0 to 2.6 h [4][5][6][7]. Hemorrhage is the most common cause of shock in the injured, and a substantial number of trauma patients will arrive at hospital with profound physiologic disturbances due to acute circulatory failure. Dr. Samuel D Gross, regarded as one of the most innovative and influential surgeons of the 19th century described shock simply as, “… a rude unhinging of the machinery of life”. Indeed, this remarkable characterization of hemorrhagic shock remains as informative today as certainly it was over 175 years ago [8].
The polytrauma victim with significant hemorrhage suffers a life-threatening acute reduction in oxygen delivery (DO 2) to tissue. DO 2 depends on both an adequate circulating blood volume representing sufficient oxygen carrying capacity, and effective cardiovascular function to maintain the circulation of blood to capillary beds in the periphery.
Furthermore, between 25% to 35% of hemorrhaging patients will develop a biochemically evident coagulopathy (trauma-induced coagulopathy; TIC) before arrival to the emergency department, which can manifest clinically as either hypercoagulable or hypocoagulable states, or both. In the polytrauma patient the presence of TIC is associated with higher transfusion requirements, increased I.C.U. and hospital length of stay (LOS), prolonged requirement for mechanical ventilation, an increase in the incidence of multiorgan dysfunction, and, most concerning of all, a threefold to fourfold higher rate of mortality [9][10][11][12][13]. TIC has deleterious effects independent of injury severity, level of shock, degree of acidosis or depth of hypothermia [14].
2. Pathophysiology of Hemorrhagic Sock
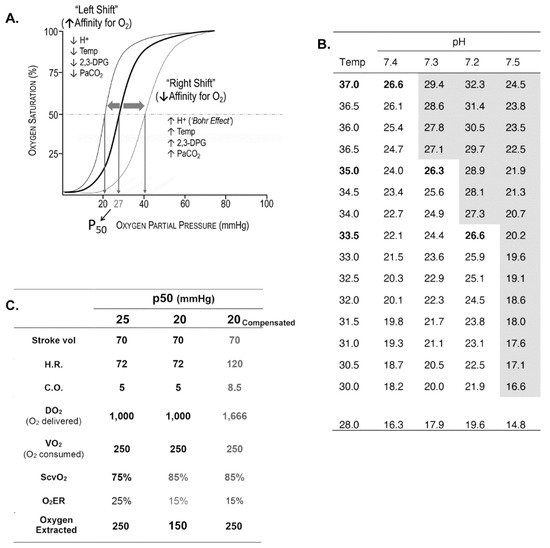
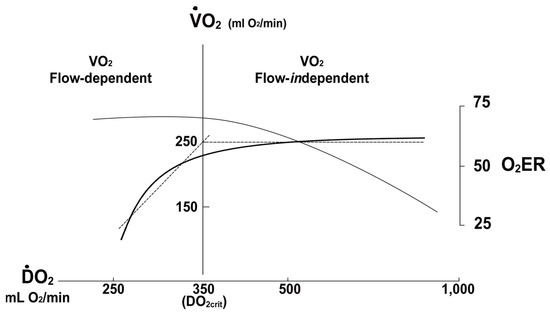
Oxygen Delivery/Utilization Imbalance
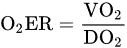
References
- Demetriades, D.; Murray, J.; Martin, M.; Velmahos, G.; Salim, A.; Alo, K.; Rhee, P. Pedestrians injured by automobiles: Relationship of age to injury type and severity. J. Am. Coll. Surg. 2004, 199, 382–387.
- Moore, F.A.; Nelson, T.; McKinley, B.A.; Moore, E.E.; Nathens, A.B.; Rhee, P.; Puyana, J.C.; Beilman, G.J.; Cohn, S.M.; StO2 Study Group. Is there a role for aggressive use of fresh frozen plasma in massive transfusion of civilian trauma patients? Am. J. Surg. 2008, 196, 948–958; discussion 958–960.
- Cannon, J.W. Hemorrhagic Shock. N. Engl. J. Med. 2018, 378, 370–379.
- Tisherman, S.A.; Schmicker, R.H.; Brasel, K.J.; Bulger, E.M.; Kerby, J.D.; Minei, J.P.; Powell, J.L.; Reiff, D.A.; Rizoli, S.B.; Schreiber, M.A. Detailed description of all deaths in both the shock and traumatic brain injury hypertonic saline trials of the Resuscitation Outcomes Consortium. Ann. Surg. 2015, 261, 586–590.
- Hauser, C.J.; Boffard, K.; Dutton, R.; Bernard, G.R.; Croce, M.A.; Holcomb, J.B.; Leppaniemi, A.; Parr, M.; Vincent, J.L.; Tortella, B.J.; et al. Results of the CONTROL trial: Efficacy and safety of recombinant activated Factor VII in the management of refractory traumatic hemorrhage. J. Trauma 2010, 69, 489–500.
- Holcomb, J.B.; del Junco, D.J.; Fox, E.E.; Wade, C.E.; Cohen, M.J.; Schreiber, M.A.; Alarcon, L.H.; Bai, Y.; Brasel, K.J.; Bulger, E.M.; et al. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: Comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013, 148, 127–136.
- Fox, E.E.; Holcomb, J.B.; Wade, C.E.; Bulger, E.M.; Tilley, B.C.; Group, P.S. Earlier Endpoints are Required for Hemorrhagic Shock Trials Among Severely Injured Patients. Shock 2017, 47, 567–573.
- Mullins, R.J.; Trunkey, D.D. Samuel, D. Gross: Pioneer academic trauma surgeon of 19th century America. J. Trauma 1990, 30, 528–538.
- Maegele, M.; Lefering, R.; Yucel, N.; Tjardes, T.; Rixen, D.; Paffrath, T.; Simanski, C.; Neugebauer, E.; Bouillon, B.; AG Polytrauma of the German Trauma Society (DGU). Early coagulopathy in multiple injury: An analysis from the German Trauma Registry on 8724 patients. Injury 2007, 38, 298–304.
- MacLeod, J.B.; Lynn, M.; McKenney, M.G.; Cohn, S.M.; Murtha, M. Early coagulopathy predicts mortality in trauma. J. Trauma 2003, 55, 39–44.
- Niles, S.E.; McLaughlin, D.F.; Perkins, J.G.; Wade, C.E.; Li, Y.; Spinella, P.C.; Holcomb, J.B. Increased mortality associated with the early coagulopathy of trauma in combat casualties. J. Trauma 2008, 64, 1459–1463; discussion 1463–1465.
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Matthay, M.A.; Mackersie, R.C.; Pittet, J.F. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann. Surg. 2007, 245, 812–818.
- Brohi, K.; Singh, J.; Heron, M.; Coats, T. Acute traumatic coagulopathy. J. Trauma 2003, 54, 1127–1130.
- Kutcher, M.E.; Howard, B.M.; Sperry, J.L.; Hubbard, A.E.; Decker, A.L.; Cuschieri, J.; Minei, J.P.; Moore, E.E.; Brownstein, B.H.; Maier, R.V.; et al. Evolving beyond the vicious triad: Differential mediation of traumatic coagulopathy by injury, shock, and resuscitation. J. Trauma Acute Care Surg. 2015, 78, 516–523.
- Ekeloef, N.P.; Eriksen, J.; Kancir, C.B. Evaluation of two methods to calculate p50 from a single blood sample. Acta Anaesthesiol. Scand. 2001, 45, 550–552.
- Srinivasan, A.J.; Morkane, C.; Martin, D.S.; Welsby, I.J. Should modulation of p50 be a therapeutic target in the critically ill? Expert Rev. Hematol. 2017, 10, 449–458.
- Leach, R.M.; Treacher, D.F. The pulmonary physician in critical care * 2: Oxygen delivery and consumption in the critically ill. Thorax 2002, 57, 170–177.
- Abdelsalam, M.; Cheifetz, I.M. Goal-directed therapy for severely hypoxic patients with acute respiratory distress syndrome: Permissive hypoxemia. Respir. Care 2010, 55, 1483–1490.
- Mercado, P.; Maizel, J.; Beyls, C.; Titeca-Beauport, D.; Joris, M.; Kontar, L.; Riviere, A.; Bonef, O.; Soupison, T.; Tribouilloy, C.; et al. Transthoracic echocardiography: An accurate and precise method for estimating cardiac output in the critically ill patient. Crit. Care 2017, 21, 136.
- Jozwiak, M.; Monnet, X.; Teboul, J.L. Monitoring: From cardiac output monitoring to echocardiography. Curr. Opin. Crit. Care 2015, 21, 395–401.
- Cecconi, M.; De Backer, D.; Antonelli, M.; Beale, R.; Bakker, J.; Hofer, C.; Jaeschke, R.; Mebazaa, A.; Pinsky, M.R.; Teboul, J.L.; et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014, 40, 1795–1815.
- Antonelli, M.; Levy, M.; Andrews, P.J.; Chastre, J.; Hudson, L.D.; Manthous, C.; Meduri, G.U.; Moreno, R.P.; Putensen, C.; Stewart, T.; et al. Hemodynamic monitoring in shock and implications for management. International Consensus Conference, Paris, France, 27–28 April 2006. Intensive Care Med. 2007, 33, 575–590.
- Narang, N.; Thibodeau, J.T.; Levine, B.D.; Gore, M.O.; Ayers, C.R.; Lange, R.A.; Cigarroa, J.E.; Turer, A.T.; de Lemos, J.A.; McGuire, D.K. Inaccuracy of estimated resting oxygen uptake in the clinical setting. Circulation 2014, 129, 203–210.
- Lubarsky, D.A.; Smith, L.R.; Sladen, R.N.; Mault, J.R.; Reed, R.L., 2nd. Defining the relationship of oxygen delivery and consumption: Use of biologic system models. J. Surg. Res. 1995, 58, 503–508.


