Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yuri V. Sergeev | + 2512 word(s) | 2512 | 2021-09-28 08:05:30 | | | |
| 2 | Amina Yu | -231 word(s) | 2281 | 2021-10-29 05:45:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sergeev, Y.V. Tyrp1 Mutant Variants and OCA3. Encyclopedia. Available online: https://encyclopedia.pub/entry/15529 (accessed on 08 February 2026).
Sergeev YV. Tyrp1 Mutant Variants and OCA3. Encyclopedia. Available at: https://encyclopedia.pub/entry/15529. Accessed February 08, 2026.
Sergeev, Yuri V.. "Tyrp1 Mutant Variants and OCA3" Encyclopedia, https://encyclopedia.pub/entry/15529 (accessed February 08, 2026).
Sergeev, Y.V. (2021, October 29). Tyrp1 Mutant Variants and OCA3. In Encyclopedia. https://encyclopedia.pub/entry/15529
Sergeev, Yuri V.. "Tyrp1 Mutant Variants and OCA3." Encyclopedia. Web. 29 October, 2021.
Copy Citation
Oculocutaneous albinism type 3 (OCA3) is an autosomal recessive disorder caused by mutations in the TYRP1 gene. Tyrosinase-related protein 1 (Tyrp1) is involved in eumelanin synthesis, catalyzing the oxidation of 5,6-dihydroxyindole-2-carboxylic acid oxidase (DHICA) to 5,6-indolequinone-2-carboxylic acid (IQCA).
melanogenesis
Tyrp1
OCA3
disease-related mutant variants
molecular modeling
1. Introduction
Human tyrosinase-related protein 1 (Tyrp1) is a transmembrane, metal-containing glycoenzyme that catalyzes the oxidation of 5,6-dihydroxyindole-2-carboxylic acid (DHICA) to 5,6-quinone-2-carboxylic acid (IQCA). It is one of three tyrosinase-like enzymes in human melanocytes that are involved in the biosynthesis of melanin, a pigment found in hair, skin, and the iris of the eye. Mutations in the Tyrp1 gene (TYRP1) can lead to oculocutaneous albinism type 3 (OCA3), an autosomal recessive disease. Those with OCA3 typically present with one of two phenotypes: rufous OCA (ROCA) or brown OCA (BOCA). ROCA is characterized by red-bronze skin color, blue or brown irises, and ginger-red hair, while BOCA is characterized by light to brown hair and light to brown or tan skin color.
Four conserved regions exist among all three tyrosinase-like enzymes: an N-terminal signal peptide, an intra-melanosomal domain, one transmembrane α-helix, and a small C-terminal cytoplasmic domain. Within the intra-melanosomal domain, there exist both a Cys-rich subdomain and a tyrosinase-like subdomain containing a binuclear metal-ion binding motif. The crystal structure of the intra-melanosomal fragment of Tyrp1, which includes both subdomains (residues 25–470) was determined at 2.35 Å [1]. In total, Tyrp1 contains 537 residues comprising the signal peptide (1–24), the intra-melanosomal domain (Cys-rich subdomain 25–126, tyrosinase-like subdomain 127–477), the transmembrane α-helix (478–501), and the cytoplasmic domain (502–537).
The core of the Cys-rich subdomain of Tyrp1 resembles the epidermal growth factor (EGF)-like domain due to its patterned disulfide bridges and short antiparallel ß-strands [1][2]. It contains five total disulfide bridges, three of them located within the EGF-like fold. Most albinism-related mutations of Tyrp1 affect its stability or activity, and expansion of the interface between the EGF-like domain and the tyrosinase-like domain was linked to OCA Type 1B mutations in tyrosinase [1][3]. One such mutation, C30R, breaks the C30-C41 disulfide bridge and results in OCA3 [4]. Tyrp1 was found to have a binuclear zinc active site rather than the typical binuclear copper active site found in tyrosinases (Figure 1).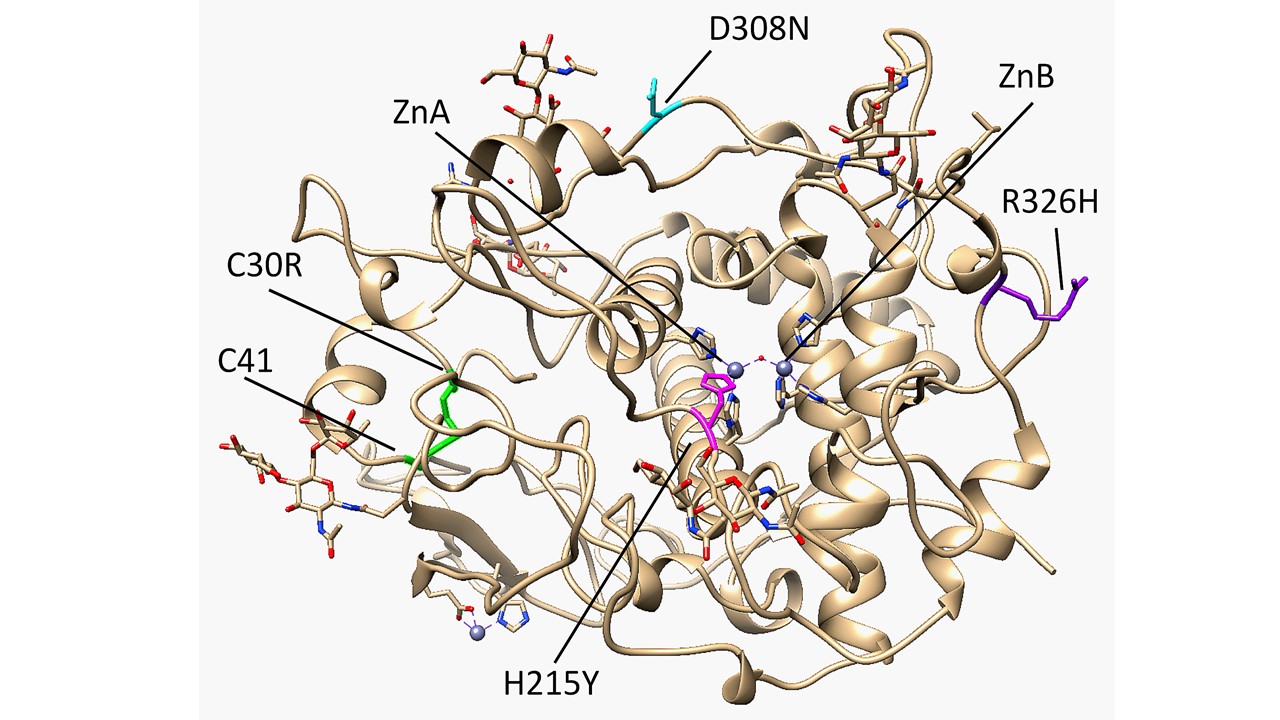 A four-helix bundle within the tyrosinase-like subdomain contains six total histidine residues that coordinate the Zn metal ions, which are bridged by a single water molecule. ZnA is coordinated by H192, H215, and H224, while ZnB is coordinated by H377, H381, and H404. In tyrosinase, histidine is essential for catalytic activity, and in both tyrosinase and Tyrp1, mutations to metal-coordinating histidine residues can result in reduced activity and consequently different forms of albinism [5][6]. Mutations to H215, H224, and H381 were specifically identified to have OCA3 [7][8]. Tyrp1 is an N-linked glycoprotein that contains sugars at six N sites (96, 104, 181, 304, 350, 385). Glycosylation has been shown to help ensure proper folding and stabilize tyrosinase-like structures during the process of translocation from the endoplasmic reticulum (ER) to the cytoplasm of the melanocyte [6][9][10].
A four-helix bundle within the tyrosinase-like subdomain contains six total histidine residues that coordinate the Zn metal ions, which are bridged by a single water molecule. ZnA is coordinated by H192, H215, and H224, while ZnB is coordinated by H377, H381, and H404. In tyrosinase, histidine is essential for catalytic activity, and in both tyrosinase and Tyrp1, mutations to metal-coordinating histidine residues can result in reduced activity and consequently different forms of albinism [5][6]. Mutations to H215, H224, and H381 were specifically identified to have OCA3 [7][8]. Tyrp1 is an N-linked glycoprotein that contains sugars at six N sites (96, 104, 181, 304, 350, 385). Glycosylation has been shown to help ensure proper folding and stabilize tyrosinase-like structures during the process of translocation from the endoplasmic reticulum (ER) to the cytoplasm of the melanocyte [6][9][10].
A four-helix bundle within the tyrosinase-like subdomain contains six total histidine residues that coordinate the Zn metal ions, which are bridged by a single water molecule. ZnA is coordinated by H192, H215, and H224, while ZnB is coordinated by H377, H381, and H404. In tyrosinase, histidine is essential for catalytic activity, and in both tyrosinase and Tyrp1, mutations to metal-coordinating histidine residues can result in reduced activity and consequently different forms of albinism [5][6]. Mutations to H215, H224, and H381 were specifically identified to have OCA3 [7][8]. Tyrp1 is an N-linked glycoprotein that contains sugars at six N sites (96, 104, 181, 304, 350, 385). Glycosylation has been shown to help ensure proper folding and stabilize tyrosinase-like structures during the process of translocation from the endoplasmic reticulum (ER) to the cytoplasm of the melanocyte [6][9][10].The unfolding mutation screen (UMS) was developed to understand and evaluate the effect of missense mutations on protein folding and thermodynamic stability [11][12]. This program uses protein unfolding curves and thermodynamic changes in Gibbs free energy (ΔΔG), determined by FoldX to calculate propensities of mutations in global mutagenesis [13]. Initially, UMS [11] was tested with 16 crystal structures to evaluate the unfolding of 1391 mutations from the ProTherm database. UMS showed that the computational accuracy of the unfolding calculations was similar to the accuracy of previously published free energy changes, but provided a better scale. The results are then projected onto a protein model to highlight the critical residues and regions of structural importance to the protein [14].
While mutations leading to OCA3 have been discovered, the molecular mechanisms leading to these results have yet to be elucidated. Here, for the first time, we used computational methods to identify mechanisms behind mutation instability and differences behind protein-ligand interactions in Tyrp1 and OCA3-causing mutations. Understanding the differences in protein-ligand interactions among Tyrp1 and its associated mutations can establish mechanisms by which they change Tyrp1 catalytic activity. Coupled with in vitro and in vivo studies, in silico studies of similar nature can help lead to novel therapies for OCA3 patients.
2. Tyrp1 Mutant Variants Associated with OCA3: Computational Characterization of Protein Stability and Ligand Binding
Tyrp1 is one of three key enzymes involved in the melanogenesis pathway, which results in the production of melanin pigment. It is a glycoenzyme that catalyzes the oxidation of DHICA to IQCA. Mutations to the TYRP1 gene can result in the autosomal recessive disorder OCA3. Four OCA3 mutants of interest were chosen from Human Gene Mutation Database (HGMD) Professional | QIAGEN to better understand Tyrp1 protein stability and catalytic activity: C30R, H215Y, D308N, and R326H. Global mutagenesis and Western Blot suggested that C30R and H215Y were severely unstable, while D308N and R326H conserved some level of catalytic activity. Free energy landscapes of the Tyrp1 bottleneck demonstrated that a slight loss of catalytic activity in D308N and R326H may be due to a larger active site, affecting interactions that stabilize DHICA during catalysis. These non-covalent interactions include salt bridges from D212 and E216 as well as various hydrogen bonds. Structural perturbations resulting from the D308N and R326H mutants may result in allosteric alterations to the active site that affect the stability of the ligand during the redox reaction.
UMS predicted mutants C30R and H215Y would be completely misfolded or unfolded, and mutants D308N and R326H to retain some stability. The results of global mutagenesis were confirmed by experimental SDS-PAGE and Western Blot, providing further validity to UMS and its ability to predict mutant stability using the foldability parameter.
C30R is in the Cys-rich subdomain, and it breaks the C30-C41 disulfide bridge. The Cys-rich subdomain of Tyrp1 is an extremely stable portion of the protein and a loss of a disulfide bridge may introduce thermodynamic instability and structural flexibility to this region. While the simulation did not suggest mutant unfolding, the mutation could alter the folding process while C30R is synthesized in the ER, resulting in a completely misfolded or unfolded protein.
H215Y is located within the active site, and it removes one of three coordination sites for ZnA. UMS also expects mutations of H215 to have a severe effect on the structural and thermodynamic stability of Tyrp1, and the ∆∆G value of H215Y indicates the greatest effect out of all four mutants of interest. The expression and purification of H215Y are consistent with the results generated by UMS. The loss of a coordination bond is expected to affect active site stability, particularly the zinc atom’s ability to remain situated within the binding pocket, resulting in a lack of catalytic activity. A comparison of H215Y structures at various points in the simulation did not suggest any active site structural alterations. All four transmembrane helices and the zinc-water-zinc complex remained in place. Residue-residue distance maps did not suggest structural fluctuations not seen in Tyrp1. However, since the mutant is expected to be unfolded or misfolded, the lack of trends towards structural alterations indicates that the protein may be unable to properly complete the folding process as it is synthesized in the ER. Similar results were seen in copper-coordinating histidine OCA1A mutations in human tyrosinase (Tyr) [3]. It was suggested that the folding of Tyr occurs in four distinct stages. Folding of the Cys-rich region and folding of each half of the transmembrane helix bundle comprise the first three stages, with the latter two stages forming the active site. Similar to Tyr, the Tyrp1 helix bundle is comprised of two helix-loop-helix motifs. Each motif contains three zinc-coordinating histidine residues and is small enough to form within a ribosome exit tunnel. Once the entire protein is processed by the ribosome, both helix-loop-helix motifs come together with the zinc-water-zinc complex to form the active site. However, without all six zinc-coordinating histidine residues, the zinc atoms are unable to correctly position themselves within the active site, and the transmembrane helix bundle does not form, leading to a full loss of catalytic activity in Tyrp1 due to denaturation.
D308N is located on the periphery of the protein, and it introduces the formation of two transient hydrogen bonds. D308 is not expected to be critical to Tyrp1 stability, and UMS does not expect D308N to be unfolded or misfolded, having the lowest ∆∆G of any of the four mutants. The expression and purification of D308N are consistent with UMS results. The addition of hydrogen bonds suggests that the stability of the mutant region might be increased. Effects of the mutation may result in allosteric alterations to active site structure as described by the most probable conformations of the bottleneck residues. The size of the bottleneck increases compared to Tyrp1, and computational docking experiments indicate that this difference may affect ligand-receptor interactions. While the number and type of noncovalent interactions between DHICA and D308N remain similar to those of Tyrp1, the strength of the stabilizing salt bridges decreases due to distance. The increased distances of these interactions are reflected in the surface images, where DHICA is not as tightly bound to the active site, leading to a loss of catalytic activity in D308N.
R326H is also located on the periphery of the protein, and it has two transient hydrogen bonds with E329. Mutations to R326 are expected to have varying levels of influence on Tyrp1 stability, and UMS does not expect R326H to be completely unfolded or misfolded. The expression and purification of R326H are consistent with UMS results. The removal of two hydrogen bonds suggests some loss of stability, and like D308N, this change in protein stability may result in allosteric alterations to the bottleneck structure. Again, the bottleneck increases compared to Tyrp1′s bottlenecks, but unlike D308N, the free energy landscapes do not indicate any centralized peak, and the altered bottleneck affects ligand-receptor interactions. The total number of noncovalent interactions decreases, subsequently decreasing the stability of the ligand in the active site. Fewer interactions are reflected in the surface images, where large gaps appear between DHICA and R326H. These gaps indicate the DHICA is not as tightly bound to the protein, leading to a loss of catalytic activity in R326H.
While both the D308N and R326H mutations do not directly affect the stability of the active site, their allosteric impacts may result in free energy changes. Enzymes have been shown to have residues that are part of allosteric regulatory sites critical to long-range communication [15]. A conservative replacement of D308 and R326 could result in free energy alterations that affect protein-ligand interactions. Both mutations are within 15 Å of transmembrane helices containing zinc-coordinating histidine residues, and mutational perturbations could affect the packing and dynamics of these helices that comprise the active site. Previous studies have shown that this long-range modulation of protein features can alter protein stability, thereby contributing to allosteric modulation of function [16]. Both D308 and R326 may be part of an intra-protein interaction network that allosterically affects the structure of the active site, particularly the bottleneck residues. This allosteric effect could be interpreted at the level of interatomic interactions. The addition and removal of hydrogen bonds in D308N and R326H, respectively, could perturb the transmembrane helices, increasing the size of the bottleneck and weakening the interactions between DHICA and the protein. These weakened interactions, in turn, affect the stabilization of DHICA within the active site and could result in reduced catalytic activity. In both the Tyrp1 and R326H docking experiments, DHICA was shown to accept a hydrogen bond from the active site water molecule. While previous studies indicated that the water molecule’s purpose was to bridge the zinc atoms, it may serve multiple functions within the active site, including stabilization of the DHICA ligand.
Although UMS foldability has shown to be an accurate predictor of protein expression and stability, it remains to be seen if this translates to phenotypic variations in OCA3. Since D308N and R326H are expected to have some level of protein activity, patients with these mutations should have reduced the severity of OCA3. If UMS can accurately predict the extent of these mutations, it could be a useful resource in helping guide personal patient therapies to those affected by OCA3.
3. Conclusions
For the first time, the mutant variants of Tyrp1, C30R, H215Y, D308N, and R326H were analyzed computationally. We have demonstrated that the protein stability changes caused by the disease-causing mutation could be predicted using the unfolding mutation screen. For three mutant variants, the predicted changes are associated with OCA3 clinical data on genetic disease severity. We demonstrated that both the C30R and H215Y mutants result in a complete loss of protein stability as they affect structurally integral regions of Tyrp1, which is consistent with in vitro experiments. Both D308N and R326H conserve the structure of these regions but may allosterically affect the conformation of the active site, increasing the size of the bottleneck. This effect is highlighted in protein-ligand interactions. While the noncovalent interactions largely remain similar between Tyrp1 and D308N, their strength decreases due to distance. R326H, however, is shown to have an even greater effect on the bottleneck, resulting in both fewer and weaker interactions with DHICA. Identifying the bottleneck residues of active sites can help identify their most stable conformations and elucidate protein-ligand interactions integral to catalytic activity. These studies, coupled with in vitro and in vivo experiments, can advance our understanding of the mechanisms behind mutations of Tyrp1 and lead to novel therapies for patients with OCA3.
References
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew. Chem. Int. Ed. Engl. 2017, 56, 9812–9815.
- Lu, H.-S.; Chai, J.-J.; Li, M.; Huang, B.-R.; He, C.-H.; Bi, R.-C. Crystal Structure of Human Epidermal Growth Factor and Its Dimerization. J. Biol. Chem. 2001, 276, 34913–34917.
- Patel, M.; Sergeev, Y. Functional in silico analysis of human tyrosinase and OCA1 associated mutations. J. Anal. Pharm. Res. 2020, 9, 81–89.
- Yamada, M.; Sakai, K.; Hayashi, M.; Hozumi, Y.; Abe, Y.; Kawaguchi, M.; Ihn, H.; Suzuki, T. Oculocutaneous albinism type 3: A Japanese girl with novel mutations in TYRP1 gene. J. Dermatol. Sci. 2011, 64, 217–222.
- Noh, H.; Lee, S.J.; Jo, H.-J.; Choi, H.W.; Hong, S.; Kong, K.-H. Histidine residues at the copper-binding site in human tyrosinase are essential for its catalytic activities. J. Enzym. Inhib. Med. Chem. 2020, 35, 726–732.
- Dolinska, M.B.; Kus, N.J.; Farney, S.K.; Wingfield, P.T.; Brooks, B.P.; Sergeev, Y.V. Oculocutaneous albinism type 1: A link between mutations, tyrosinase conformational stability, and enzymatic activity. Pigment. Cell Melanoma Res. 2017, 30, 41–52.
- Zhang, K.H.; Li, Z.; Lei, J.; Pang, T.; Xu, B.; Jiang, W.Y.; Li, H.Y. Oculocutaneous albinism type 3 (OCA3): Analysis of two novel mutations in TYRP1 gene in two Chinese patients. Cell Biochem. Biophys. 2011, 61, 523–529.
- Lasseaux, E.; Plaisant, C.; Michaud, V.; Pennamen, P.; Trimouille, A.; Gaston, L.; Monfermé, S.; Lacombe, D.; Rooryck, C.; Morice-Picard, F.; et al. Molecular characterization of a series of 990 index patients with albinism. Pigment. Cell Melanoma Res. 2018, 31, 466–474.
- Branza-Nichita, N.; Negroiu, G.; Petrescu, A.-J.; Garman, E.; Platt, F.; Wormald, M.; Dwek, R.A.; Petrescu, S. Mutations at Critical N-Glycosylation Sites Reduce Tyrosinase Activity by Altering Folding and Quality Control. J. Biol. Chem. 2000, 275, 8169–8175.
- Branza-Nichita, N.; Petrescu, A.J.; Negroiu, G.; Dwek, R.A.; Petrescu, S. N-Glycosylation Processing and Glycoprotein Folding−Lessons from the Tyrosinase-Related Proteins. Chem. Rev. 2000, 100, 4697–4712.
- McCafferty, C.L.; Sergeev, Y.V. In silico Mapping of Protein Unfolding Mutations for Inherited Disease. Sci. Rep. 2016, 6, 37298.
- NEI Commons Website. Ocular Proteome Webpage, National Eye Institute. 2017. Available online: https://neicommons.nei.nih.gov/#/proteomeData (accessed on 2 July 2021).
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388.
- McCafferty, C.L.; Sergeev, Y.V. Global computational mutagenesis provides a critical stability framework in protein structures. PLoS ONE 2017, 12, e0189064.
- Tyukhtenko, S.; Rajarshi, G.; Karageorgos, I.; Zvonok, N.; Gallagher, E.S.; Huang, H.; Vemuri, K.; Hudgens, J.W.; Ma, X.; Nasr, M.L.; et al. Effects of Distal Mutations on the Structure, Dynamics and Catalysis of Human Monoacylglycerol Lipase. Sci. Rep. 2018, 8, 1719.
- Naganathan, A.N. Modulation of allosteric coupling by mutations: From protein dynamics and packing to altered native ensembles and function. Curr. Opin. Struct. Biol. 2018, 54, 1–9.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
29 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No