 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giorgio Costagliola | + 1373 word(s) | 1373 | 2021-10-19 09:56:55 | | | |
| 2 | Vicky Zhou | Meta information modification | 1373 | 2021-10-29 03:35:07 | | | | |
| 3 | Vicky Zhou | + 12 word(s) | 1385 | 2021-10-29 03:39:27 | | |
Video Upload Options
Primary immunodeficiency disorders (PIDs) patients can develop an immune dysregulation of variable degree, which is responsible for a clinical picture featured by infectious complications and autoimmunity. Autoimmune manifestations are observed with considerable frequency in patients with primary antibody deficiencies, including common variable immunodeficiency (CVID) and selective IgA deficiency (sIgAD), but can also be evidenced in patients with combined immunodeficiency disorders (CID).
1. Introduction
In recent years, the association between primary immunodeficiency disorders (PIDs) and autoimmunity has been extensively studied. Notably, autoimmunity can represent the presentation sign of PIDs in a significant number of patients [1]. The molecular mechanisms responsible for the immune dysregulation in patients with PIDs are multiple and not completely elucidated; impaired B cell differentiation and germ-center reactions, altered T cell central or peripheral tolerance, uncontrolled lymphocyte proliferation and differentiation, dysfunctional complement, and innate immune activation can participate in the complex pathogenic process leading to autoimmunity. In patients with PIDs, the association with autoimmunity leads to a significant impact on the quality of life, higher medicalization, and increased mortality [2]. Furthermore, the increasing use of new sequencing techniques allowed the identification of different monogenic causes of PID, the better understanding of genotype-phenotype correlations, and the improvement of the therapeutic strategies targeting the immune dysregulation in PIDs [3][4].
2. Clinical implications
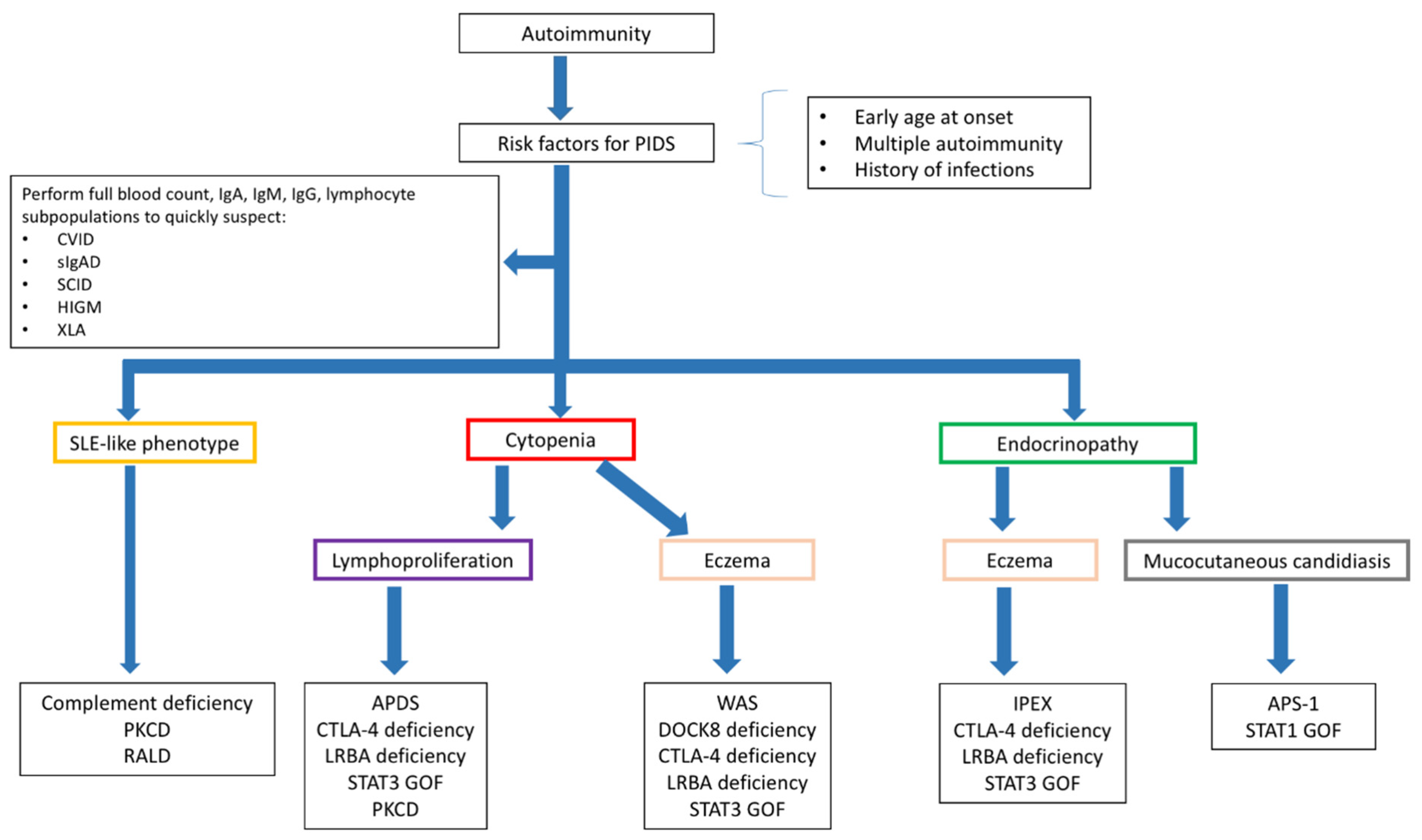
2.1. Diagnosing PIDs in Children Presenting with Autoimmunity
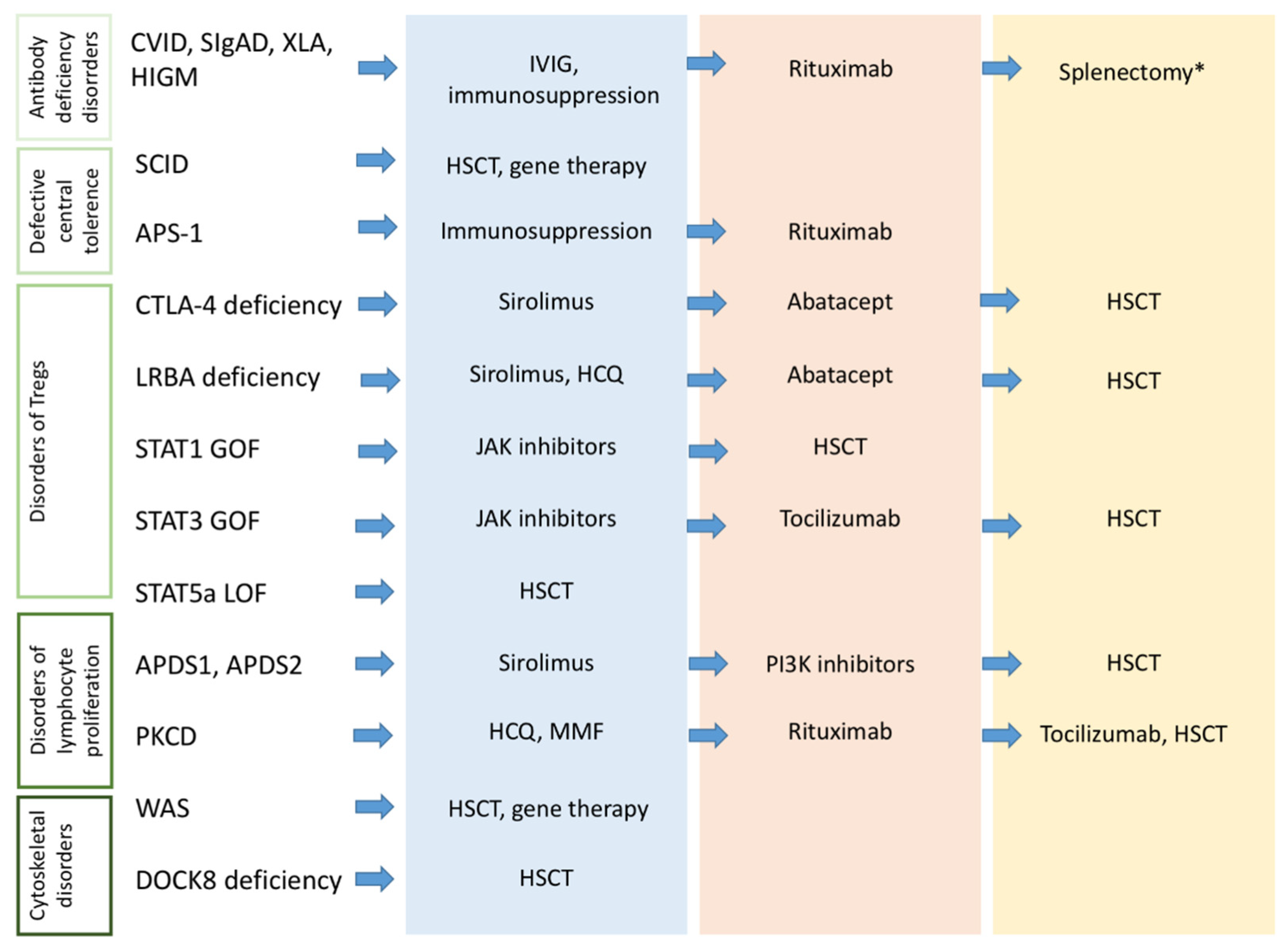
2.2. Diagnosing Autoimmunity in Children with PIDs
3. Conclusions
References
- Walter, J.E.; Ayala, I.A.; Milojevic, D. Autoimmunity as a continuum in primary immunodeficiency. Curr. Opin. Pediatr. 2019, 31, 851–862.
- Amaya-Uribe, L.; Rojas, M.; Azizi, G.; Anaya, J.M.; Gershwin, M.E. Primary immunodeficiency and autoimmunity: A comprehensive review. J. Autoimmun. 2019, 99, 52–72.
- Notarangelo, L.D.; Uzel, G.; Rao, V.K. Primary immunodeficiencies: Novel genes and unusual presentations. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 443–448.
- Bousfiha, A.; Jeddane, L.; Picard, C.; Ailal, F.; Bobby Gaspar, H.; Al-Herz, W. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J. Clin. Immunol. 2018, 38, 129–143.
- Azizi, G.; Ziaee, V.; Tavakol, M.; Alinia, T.; Yazdai, R.; Mohammadi, H.; Abolhassani, H.; Aghamohammadi, A. Approach to the Management of Autoimmunity in Primary Immunodeficiency. Scand. J. Immunol. 2017, 85, 13–29.
- Walter, J.E.; Farmer, J.R.; Foldvari, Z.; Torgerson, T.R.; Cooper, M.A. Mechanism-Based Strategies for the Management of Autoimmunity and Immune Dysregulation in Primary Immunodeficiencies. J. Allergy Clin. Immunol. Pract. 2016, 4, 1089–1100.
- Abraham, R.S. How to evaluate for immunodeficiency in patients with autoimmune cytopenias: Laboratory evaluation for the diagnosis of inborn errors of immunity associated with immune dysregulation. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 661–672.
- Consolini, R.; Costagliola, G.; Spatafora, D. The Centenary of Immune Thrombocytopenia-Part 2: Revising Diagnostic and Therapeutic Approach. Front Pediatr. 2017, 5, 179.
- Fischer, A.; Provot, J.; Jais, J.P.; Alcais, A.; Mahlaoui, N. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2017, 140, 1388–1393.e8.
- Besnard, C.; Levy, E.; Aladjidi, N.; Stolzenberg, M.C.; Magerus-Chatinet, A.; Alibeu, O.; Nitschke, P.; Blanche, S.; Hermine, O.; Jeziorski, E.; et al. Pediatric-onset Evans syndrome: Heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin. Immunol. 2018, 188, 52–57.
- Hadjadj, J.; Aladjidi, N.; Fernandes, H.; Leverger, G.; Magérus-Chatinet, A.; Mazerolles, F.; Hussey, A.A.; Evbuomwan, M.O.; Long Priel, D.A.; Kuhns, D.B.; et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. J. Am. Soc. Hematol. 2019, 134, 9–21.
- Costagliola, G.; Marco, S.D.; Comberiati, P.; D’Elios, S.; Petashvili, N.; Di Cicco, M.E.; Peroni, D. Practical Approach to Children Presenting with Eosinophila and Hypereosinophilia. Curr. Pediatr. Rev. 2020, 16, 81–88.
- Shamriz, O.; Tal, Y.; Talmon, A.; Nahum, A. Chronic Mucocutaneous Candidiasis in Early Life: Insights Into Immune Mechanisms and Novel Targeted Therapies. Front Immunol. 2020, 11, 593289.
- Leffler, J.; Bengtsson, A.A.; Blom, A.M. The complement system in systemic lupus erythematosus: An update. Ann. Rheum. Dis. 2014, 73, 1601–1606.
- Boileau, J.; Mouillot, G.; Gerard, L.; Carmagnat, M.; Rabian, C.; Oksenhendler, E.; Pasquali, J.-L.; Korganow, A.-S.; DEFI Study Group. Autoimmunity in common variable immunodeficiency: Correlation with lymphocyte phenotype in the French DEFI study. J. Autoimmun. 2011, 36, 25–32.
- Sánchez-Ramón, S.; Radigan, L.; Yu, J.E.; Bard, S.; Cunningham-Rundles, C. Memory B cells in common variable immunodeficiency: Clinical associations and sex differences. Clin. Immunol. 2008, 128, 314–321.
- Ahn, S.; Cunningham-Rundles, C. Role of B cells in common variable immune deficiency. Expert Rev. Clin. Immunol. 2009, 5, 557–564.
- Arumugakani, G.; Wood, P.M.; Carter, C.R. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J. Clin. Immunol. 2010, 30, 292–300.
- Abolhassani, H.; Amirkashani, D.; Parvaneh, N.; Mohammadinejad, P.; Gharib, B.; Shahinpour, S. Autoimmune phenotype in patients with common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2013, 23, 323–329.
- Picchianti Diamanti, A.; Rosado, M.M.; Scarsella, M.; Ceccarelli, S.; Laganà, B.; D’Amelio, R.; Carsetti, R. Increased serum IgM, immunodeficiency, and autoimmunity: A clinical series. Int. J. Immunopathol. Pharmacol. 2015, 28, 547–556.
- Warnatz, K.; Voll, R.E. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. 2012, 3, 210.
- Montin, D.; Marolda, A.; Licciardi, F.; Robasto, F.; Di Cesare, S.; Ricotti, E.; Ferro, F.; Scaioli, G.; Giancotta, C.; Amodio, D.; et al. Immunophenotype Anomalies Predict the Development of Autoimmune Cytopenia in 22q11.2 Deletion Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 2369–2376.
- McLean-Tooke, A.; Spickett, G.P.; Gennery, A.R. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand. J. Immunol. 2007, 66, 1–7.


