 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sona Ciernikova | + 1093 word(s) | 1093 | 2020-06-12 08:34:44 | | | |
| 2 | Catherine Yang | Meta information modification | 1093 | 2020-06-15 05:04:27 | | | | |
| 3 | Catherine Yang | -48 word(s) | 1045 | 2020-10-27 10:22:16 | | |
Video Upload Options
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive solid malignancies. The complex mechanism underlying the development and progression of PDAC includes interactions between genomic, epigenomic, and signaling pathway alterations. PDAC remains a devastating disease, despite extensive research efforts focused on the discovery of early diagnostic biomarkers and efficient therapeutic approaches, showing a high resistance to treatment that significantly contributes to poor survival. The success of epi-drugs in hematological malignancies and the robust body of evidence supporting the hypothesis about their ability to synergize with other anti-cancer drugs, sensitizing resistant tumor cells in preclinical models, have encouraged the search for these agents suitable for the future clinical management of PDAC.
1. Introduction
Although significant improvements have been achieved in the treatment of many solid tumors, PDAC remains poorly addressed. Besides the lack of reliable early biomarkers, the main causes include dense and diffused desmoplastic stroma, tumor heterogeneity with a broad mutational landscape, followed by overactivation of many signaling pathways that accumulate during disease progression. More than 70% of patients do not respond to the currently applied regimens and conventional treatment, chemotherapy, and radiotherapy offer marginal benefits. Usually, they are used as a first-line treatment for non-resectable disease [1]. Single-agent therapies using gemcitabine and its combinations, multidrug regimens such as FOLFIRINOX (the combination of folinic acid, 5-fluorouracil, irinotecan, oxaliplatin) and targeted approaches, have not improved patient survival [2]. Moreover, the failure of the first-line chemotherapy leads to an acceleration of growth, conducive to a resistant and metastatic tumor [3]. Immunotherapy has achieved a compelling success (mainly immune checkpoint inhibitors) in several cancers, including melanoma, urothelial, head and neck, or lung cancer [4]. Regarding PDAC, all immunotherapeutic efforts have failed so far. However, a combination of immunotherapy with epigenetic therapy, recently reviewed by Topper et al., represents a promising trend to bring the improvements in treatment of PDAC patients [5].
One of the new avenues in solid tumor clinical investigation is the potential of epi-drugs to modulate the sensitivity of tumors to other anticancer agents. Primary and acquired resistance to treatment, which is a hallmark of PDAC, may be a consequence of dynamic and reversible epigenetic changes that confer a fitness advantage to tumor cells, known as epigenetic plasticity [6]. DNA methyltransferase inhibitors (DNMTis) and Histone deacetylase inhibitors (HDACis) were originally approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for certain hematological malignancies. Great hopes were also placed in them for the treatment of solid tumors. However, despite robust preclinical evidence, first-generation epi-drugs have shown a low efficacy, such as the DNMTis azacytidine and decitabine and the HDACis vorinostat and romidepsin[7]. One possible explanation for their failure is that more differentiated solid tumors with terminally differentiated cells have a reduced capability for epigenetic reprogramming. Although the second-generation of epi-drugs (the DNMTis zebularine and guadecitabine and the HDACis belinostat, panobinostat, tucidinostat (chidamide), and valproic acid) have shown improved chemical properties, limited efficacy and significant side effects have been observed [7]. Recently, a new generation of selective epi-drugs is being developed; they are more specific for their targets and are entering clinical testing (Figure 1).
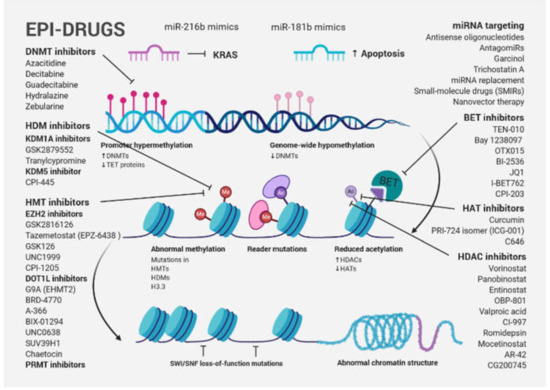
Figure 1. Schematic presentation of cancer-associated epigenetic modification abnormalities, including a brief overview of epi-drugs used in preclinical studies and clinical trials for PDAC treatment. Gene-specific hypermethylation accompanied by global DNA hypomethylation, frequent mutation of epigenetic modifiers and histone proteins, as well as abnormal histone modification patterns are typical cancer-associated epigenetic changes. A large number of the first-, second- and third-generation inhibitors, targeting epigenetic modifications, were used in preclinical studies and clinical trials. Although epi-drug monotherapies were not successful, combination therapy targeting diverse chromatin regulators or a biomarker-directed approach that combines a new generation of epi-drugs with traditional approaches currently represent the most promising concept for PDAC.
Bromodomain and extra-terminal domain inhibitors (BETis), histone methyltransferase inhibitors (HMTis), including enhancer of zeste homologue 2 inhibitors (EZH2is) and DOT1-like histone-lysine methyltransferase inhibitors (DOT1Lis), histone demethylase inhibitors (HDMis) such as lysine-specific histone demethylase 1A inhibitors (KDM1Ais) and protein arginine methyltransferase inhibitors (PRMTis), among others, are included in this group [7].
2. New Epigenetic Treatments in Pancreatic Ductal Adenocarcinoma
To reverse resistance and sensitize cancer cells to treatment, epi-drugs were tested in combination with other anticancer therapies, such as genotoxic and/or cytotoxic chemo- and radiotherapy, targeted therapies, as well as hormone and immunotherapies. Drug resistance in PDAC is mediated by pronounced cell plasticity (the ability to switch between different phenotypic states), allowing individual cellular clones to evade therapy [8]. HAT inhibitors (HATis), ICG-001 and C646, were found to reduce tumorigenicity of PDAC in preclinical cell models [9][10][11] and the PRI-724 isomer (ICG-001) is already used in a clinical trial for advanced and metastatic pancreatic adenocarcinoma (NCT01764477). However, little progress has been made to evaluate the benefit of HAT inhibition in the treatment of PDAC patients. ChIP-seq data from seven widely used PDAC cell lines corresponding to low (CAPAN1, CAPAN2, CFPAC1, HFPAC1) and high (PANC1, MiaPaCa2, PT45P1) grade groups showed that low-grade PDAC cells had an increased number of super-enhancer domains, whereas both groups had a similar number of broad H3K4me3 (a mark of active promoter regions). In the high-grade model, PANC1, treatment with either ICG-001 or C646 resulted in dramatic effects on the epigenome with hundreds of regions showing a differential enrichment of H3K27ac or H3K4me3. Interestingly, both drugs targeted similar super-enhancers, causing a reduction in histone acetylation levels near genes involved in PDAC and other solid cancers [12]. Several epi-drugs were tested in phase I/II PDAC clinical trials, namely EZH2i EPZ-6438, GSK2816126, HATi Curcumin, BETi TEN-010, Bay 1238097, OTX015 and BI-2536, and the HDACis vorinostat, panobinostat, entinostat, OBP-801, valproic acid, CI-997, romidepsin, and mocetinostat. Although epi-drug monotherapies were not successful, combination therapy that targets diverse chromatin regulators (e.g., BETis and HDACis) currently represent the most promising concept for PDAC [8]. Genes involved in cell cycle regulation (CDKN1A, XRCC6), inhibition of epithelial differentiation (GATA4, GATA6, DAB2, MUC2), apoptosis (TP53), as well as EMT and metastasis (CDH1, CXCR4, or miR-203) are HDAC targets.
The right combination, dosing, and sequence may improve the efficacy and tolerability of epi-drugs, in combination with other types of anticancer therapies. The identification of precise biomarkers for patient stratification and subsequent implementation of personalized strategies in clinical trials represent an urgent requirement for successful PDAC treatment.
References
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K.; Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141, doi:10.1186/s13014-019-1345-6.
- Adamska, A.; Domenichini, A.; Falasca, M.; Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338, doi:10.3390/ijms18071338.
- Juiz, N.A.; Iovanna, J.; Dusetti, N.; Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front. Oncol. 2019, 9, 246, doi:10.3389/fonc.2019.00246.
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al.et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268, doi:10.1186/s13046-019-1266-0.
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B.; The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75, doi:10.1038/s41571-019-0266-5.
- Grasso, C.; Jansen, G.; Giovannetti, E.; Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139, doi:10.1016/j.critrevonc.2017.03.026.
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S.; Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91, doi:10.1038/s41571-019-0267-4.
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V.; Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon?. Gut 2017, 66, 168, doi:10.1136/gutjnl-2016-312539.
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W.; The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 2014, 13, 2303, doi:10.1158/1535-7163.mct-13-1005.
- Ono, H.; Basson, M.D.; Ito, H.; P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301, doi:10.18632/oncotarget.10117.
- Manegold, P.; Lai, K.K.Y.; Wu, Y.; Teo, J.L.; Lenz, H.J.; Genyk, Y.S.; Pandol, S.J.; Wu, K.; Lin, D.P.; Chen, Y.; et al.et al. Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer. Cancers 2018, 10, 95, doi:10.3390/cancers10040095.
- Gerrard, D.L.; Boyd, J.R.; Stein, G.S.; Jin, V.X.; Frietze, S.; Disruption of Broad Epigenetic Domains in PDAC Cells by HAT Inhibitors. Epigenomes 2019, 3, 11, doi:10.3390/epigenomes3020011.


