 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lluis Espinosa | + 1435 word(s) | 1435 | 2021-09-29 09:56:38 | | | |
| 2 | Beatrix Zheng | + 38 word(s) | 1473 | 2021-10-08 12:07:31 | | |
Video Upload Options
IκBα is considered to play an almost exclusive role as inhibitor of the NF-κB signaling pathway. However, previous results have demonstrated that SUMOylation imposes a distinct subcellular distribution, regulation, NF-κB-binding affinity and function to the IκBα protein. In this review we discuss the main alterations of IκBα found in cancer and whether they are (most likely) associated with NF-κB-dependent or NF-κB-independent (moonlighting) activities of the protein.
1. IκBα Activation and Functions
The IκB proteins, including IκBα, IκBβ, IκBε, IκBζ, Bcl-3 (B-cell lymphoma 3-encoded protein), and the precursor Rel proteins p100 (IκBδ) and p105 (IκBγ), are structurally characterized by the presence of multiple ankyrin repeats, which mediate protein–protein interaction and cytoplasmic NF-κB retention. Although this family of proteins is primarily known from its almost exclusive role in canonical NF-κB inhibition [1], several NF-κB-independent functions for specific IκB subunits have been identified. Initially, it was demonstrated that physical association of IκBα to histone deacetylases (HDACs) increase NF-κB-independent transcription by cytoplasmic retention of HDAC1 and HDAC3 [2]. IκBα can also bind the chromatin at specific genomic regions to regulate gene transcription by modulation of the chromatin editing polycomb repression complex (PRC) 2 [3][4]. Mechanistically, phosphorylated and SUMOylated IκBα interacts with chromatin by direct binding to the acetylated N-terminal tail of H4 [4]. The association of IκBα to specific genes facilitates PRC2 recruitment under basal conditions but imposes the capacity of activation in response to inflammatory cytokines such as TNFα or following differentiation [4][5] ( Figure 1 ). In the same direction, it has recently been demonstrated that the dynamics of NF-κB activation can reprogram the epigenome in a stimulus specific manner [6], which may be linked to changes in IκBα protein levels.
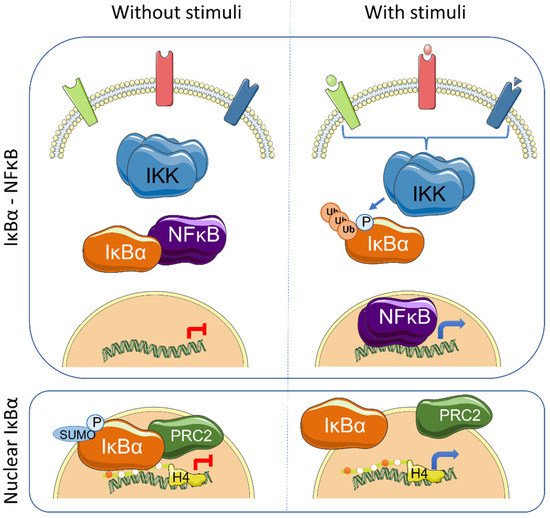
Transcriptional regulation mediated by chromatin-bound IκBα affects about 10% of all PRC2 target genes in the different models studied, and involves genes related with development, stemness and tissue homeostasis [4][5][7]. Importantly, IκBα deficiency leads to defective maturation of tissue stem cells, which are then retained in a fetal state [4][5]. The nuclear function of IκBα was first described in mammalian cells and tissues including skin [4] and intestine [5], but it is already present in organisms like Drosophila melanogaster [4] and Caenorhabditis elegans [7]. Notably, Caenorhabditis elegans , as the rest of nematodes, lacks recognizable NF-κB factors, strongly suggesting that nuclear and polycomb-related IκBα functions appeared in the evolution before or in parallel to its role as NF-κB inhibitor.
Therefore, there is cumulative data indicating that alterations related with IκBα function not only affect NF-κB pathway but IκBα exerts moonlighting functions including regulation of PRC2 activity on specific gene sets, which are pivotal for cancer initiation and progression.
2. IκBα in Hematologic Diseases
3. IκBα Loss in Glioma
4. Targeting NF-κB or/and Chromatin Editing Enzymes for Treating IκBα-Deficient Tumors
References
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel Proteins: Evolutionarily Conserved Mediators of Immune Responses. Annu. Rev. Immunol. 1998, 16, 225–260.
- Viatour, P.; Legrand-Poels, S.; van Lint, C.; Warnier, M.; Merville, M.-P.; Gielen, J.; Piette, J.; Bours, V.; Chariot, A. Cytoplasmic IκBα Increases NF-κB-independent Transcription through Binding to Histone Deacetylase (HDAC) 1 and HDAC3. J. Biol. Chem. 2003, 278, 46541–46548.
- Aguilera, C.; Hoya-Arias, R.; Haegeman, G.; Espinosa, L.; Bigas, A. Recruitment of I B to the hes1 promoter is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2004, 101, 16537–16542.
- Mulero, M.; Ferres-Marco, D.; Islam, A.; Margalef, P.; Pecoraro, M.; Toll, A.; Drechsel, N.; Charneco, C.; Davis, S.; Bellora, N.; et al. Chromatin-bound IκBα regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 2013, 24, 151–166.
- Marruecos, L.; Bertran, J.; Guillén, Y.; González, J.; Batlle, R.; López-Arribillaga, E.; Garrido, M.; Ruiz-Herguido, C.; Lisiero, D.; González-Farré, M.; et al. IκBα deficiency imposes a fetal phenotype to intestinal stem cells. EMBO Rep. 2020, 21, e49708.
- Cheng, Q.J.; Ohta, S.; Sheu, K.M.; Spreafico, R.; Adelaja, A.; Taylor, B.; Hoffmann, A. NF-κB dynamics determine the stimulus specificity of epigenomic reprogramming in macrophages. Science 2021, 372, 1349–1353.
- Brena, D.; Bertran, J.; Porta-de-la-Riva, M.; Guillén, Y.; Cornes, E.; Kukhtar, D.; Campos-Vicens, L.; Fernández, L.; Pecharroman, I.; García-López, A.; et al. Ancestral function of Inhibitors-of-kappaB regulates Caenorhabditis elegans development. Sci. Rep. 2020, 10, 1–13.
- Wood, K.M.; Roff, M.; Hay, R.T. Defective IκBα in Hodgkin cell lines with constitutively active NF-κB. Oncogene 1998, 16, 2131–2139.
- Cabannes, E.; Khan, G.; Aillet, F.; Jarrett, R.F.; Hay, R.T. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IκBα. Oncogene 1999, 18, 3063–3070.
- Jungnickel, B.; Staratschek-Jox, A.; Bräuninger, A.; Spieker, T.; Wolf, J.; Diehl, V.; Hansmann, M.-L.; Rajewsky, K.; Küppers, R. Clonal Deleterious Mutations in the Iκbα Gene in the Malignant Cells in Hodgkin’s Lymphoma. J. Exp. Med. 2000, 191, 395–402.
- Krappmann, D.; Emmerich, F.; Kordes, U.; Scharschmidt, E.; Dörken, B.; Scheidereit, C. Molecular mechanisms of constitutive NF-κB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene 1999, 18, 943–953.
- Emmerich, F.; Meiser, M.; Hummel, M.; Demel, G.; Foss, H.-D.; Jundt, F.; Mathas, S.; Krappmann, D.; Scheidereit, C.; Stein, H.; et al. Overexpression of I Kappa B Alpha Without Inhibition of NF-κB Activity and Mutations in the I Kappa B Alpha Gene in Reed-Sternberg Cells. Blood 1999, 94, 3129–3134.
- Sarkozy, C.; Hung, S.; Takata, K.; Chavez, E.; Aoki, T.; Duns, G.; Slack, G.W.; Telenius, A.; Miyata-Takata, T.; Viganò, E.; et al. Mutational Landscape of Grey Zone Lymphoma. Blood 2019, 134, 21.
- Thomas, R.K.; Wickenhauser, C.; Tawadros, S.; Diehl, V.; Küppers, R.; Wolf, J.; Schmitz, R. Mutational analysis of the I κ B α gene in activated B cell-like diffuse large B-cell lymphoma. Br. J. Haematol. 2004, 126, 50–54.
- Kalaitzidis, D.; Davis, R.E.; Rosenwald, A.; Staudt, L.M.; Gilmore, T.D. The human B-cell lymphoma cell line RC-K8 has multiple genetic alterations that dysregulate the Rel/NF-κB signal transduction pathway. Oncogene 2002, 21, 8759–8768.
- Kalaitzidis, D.; Gilmore, T.D. Genomic organization and expression of the rearrangedREL proto-oncogene in the human B-cell lymphoma cell line RC-K8. Genes Chromosom. Cancer 2002, 34, 129–135.
- Savage, K.J. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879.
- Takahashi, H.; Feuerhake, F.; Monti, S.; Kutok, J.L.; Aster, J.C.; Shipp, M.A. Lack of IKBA coding region mutations in primary mediastinal large B-cell lymphoma and the host response subtype of diffuse large B-cell lymphoma. Blood 2006, 107, 844–845.
- Crivellaro, S.; Carrà, G.; Panuzzo, C.; Taulli, R.; Guerrasio, A.; Saglio, G.; Morotti, A. The non-genomic loss of function of tumor suppressors: An essential role in the pathogenesis of chronic myeloid leukemia chronic phase. BMC Cancer 2016, 16, 314.
- Marruecos, L.; Manils, J.; Moreta, C.; Gómez, D.; Filgaira, I.; Serafin, A.; Cañas, X.; Espinosa, L.; Soler, C. Single loss of a Trp53 allele triggers an increased oxidative, DNA damage and cytokine inflammatory responses through deregulation of IκBα expression. Cell Death Dis. 2021, 12, 359.
- Zhou, M.; Gu, L.; Zhu, N.; Woods, W.G.; Findley, H.W. Transfection of a dominant-negative mutant NF-kB inhibitor (IkBm) represses p53-dependent apoptosis in acute lymphoblastic leukemia cells: Interaction of IkBm and p53. Oncogene 2003, 22, 8137–8144.
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Jain, N.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 2008, 29, 861–868.
- Parker, K.M.; Ma, M.H.; Manyak, S.; Altamirano, C.V.; Tang, Y.M.; Frantzen, M.; Mikail, A.; Roussos, E.; Sjak-Shie, N.; Vescio, R.A.; et al. Identification of polymorphisms of the IκBα gene associated with an increased risk of multiple myeloma. Cancer Genet. Cytogenet. 2002, 137, 43–48.
- Spink, C.F.; Gray, L.C.; Davies, F.E.; Morgan, G.J.; Bidwell, J.L. Haplotypic structure across the IκBα gene (NFKBIA) and association with multiple myeloma. Cancer Lett. 2007, 246, 92–99.
- Angileri, F.F.; Aguennouz, M.; Conti, A.; La Torre, D.; Cardali, S.; Crupi, R.; Tomasello, C.; Germanò, A.; Vita, G.; Tomasello, F. Nuclear factor-κB activation and differential expression of survivin and Bcl-2 in human grade 2-4 astrocytomas. Cancer 2008, 112, 2258–2266.
- Korkolopoulou, P.; Levidou, G.; Saetta, A.A.; El-Habr, E.; Eftichiadis, C.; Demenagas, P.; Thymara, I.; Xiromeritis, K.; Boviatsis, E.; Thomas-Tsagli, E.; et al. Expression of nuclear factor-κB in human astrocytomas: Relation to pIκBa, vascular endothelial growth factor, Cox-2, microvascular characteristics, and survival. Hum. Pathol. 2008, 39, 1143–1152.
- Bredel, M.; Scholtens, D.M.; Yadav, A.K.; Alvarez, A.A.; Renfrow, J.J.; Chandler, J.P.; Yu, I.L.Y.; Carro, M.S.; Dai, F.; Tagge, M.J.; et al. NFKBIA Deletion in Glioblastomas. N. Engl. J. Med. 2011, 364, 627–637.
- Kinker, G.S.; Thomas, A.M.; Carvalho, V.J.; Lima, F.P.; Fujita, A. Deletion and low expression of NFKBIA are associated with poor prognosis in lower-grade glioma patients. Sci. Rep. 2016, 6, 24160.
- Miyar, A.; Habibi, I.; Ebrahimi, A.; Mansourpour, D.; Mokarizadeh, A.; Rajabi, A.; Farshgar, R.; Eshaghzadeh, M.; Zamani-Ahmadmahmudi, M.; Nodushan, S.M.H.T. Predictive and prognostic value of TLR9 and NFKBIA gene expression as potential biomarkers for human glioma diagnosis. J. Neurol. Sci. 2016, 368, 314–317.
- Banerjee, S.; Sahoo, A.K.; Chattopadhyay, A.; Ghosh, S.S. Recombinant I κ B α -loaded curcumin nanoparticles for improved cancer therapeutics. Nanotechnology 2014, 25, 345102.
- Zhao, Z.; Zhong, X.; Wu, T.; Yang, T.; Chen, G.; Xie, X.; Wei, Y.; Ye, M.; Zhou, Y.; Du, Z. Identification of a NFKBIA polymorphism associated with lower NFKBIA protein levels and poor survival outcomes in patients with glioblastoma multiforme. Int. J. Mol. Med. 2014, 34, 1233–1240.
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40.
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; Poulaki, V.; Tai, Y.-T.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: Therapeutic applications. Blood 2003, 101, 2377–2380.
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007, 130, 918–931.
- Eich, M.-L.; Athar, M.; Ferguson, J.E.; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458.


