IκBα is considered to play an almost exclusive role as inhibitor of the NF-κB signaling pathway. However, previous results have demonstrated that SUMOylation imposes a distinct subcellular distribution, regulation, NF-κB-binding affinity and function to the IκBα protein. In this review we discuss the main alterations of IκBα found in cancer and whether they are (most likely) associated with NF-κB-dependent or NF-κB-independent (moonlighting) activities of the protein.
1. IκBα Activation and Functions
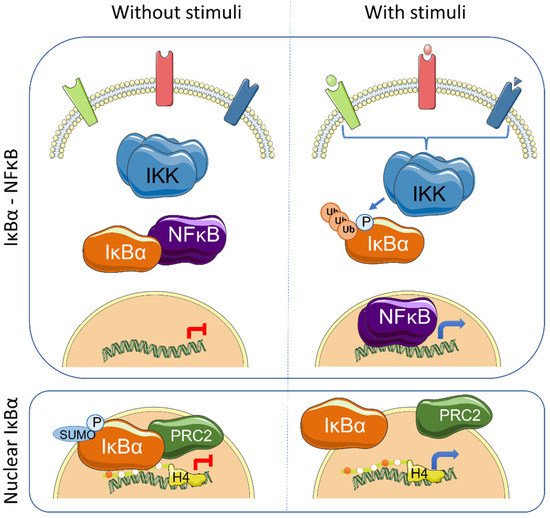
The IκB proteins, including IκBα, IκBβ, IκBε, IκBζ, Bcl-3 (B-cell lymphoma 3-encoded protein), and the precursor Rel proteins p100 (IκBδ) and p105 (IκBγ), are structurally characterized by the presence of multiple ankyrin repeats, which mediate protein–protein interaction and cytoplasmic NF-κB retention. Although this family of proteins is primarily known from its almost exclusive role in canonical NF-κB inhibition [1], several NF-κB-independent functions for specific IκB subunits have been identified. Initially, it was demonstrated that physical association of IκBα to histone deacetylases (HDACs) increase NF-κB-independent transcription by cytoplasmic retention of HDAC1 and HDAC3 [2][11]. IκBα can also bind the chromatin at specific genomic regions to regulate gene transcription by modulation of the chromatin editing polycomb repression complex (PRC) 2 [3][4][12,13]. Mechanistically, phosphorylated and SUMOylated IκBα interacts with chromatin by direct binding to the acetylated N-terminal tail of H4 [4][13]. The association of IκBα to specific genes facilitates PRC2 recruitment under basal conditions but imposes the capacity of activation in response to inflammatory cytokines such as TNFα or following differentiation [4][5][13,14] ( Figure 1 ). In the same direction, it has recently been demonstrated that the dynamics of NF-κB activation can reprogram the epigenome in a stimulus specific manner [6][15], which may be linked to changes in IκBα protein levels.
Figure 1. Graphical representation of the main IκBα-regulated pathways: NF-κB and nuclear SUMOylated IκBα. Left panels show the status of the involved elements under unstimulated conditions whereas right panels indicate changes produced after cytokine induction.
Transcriptional regulation mediated by chromatin-bound IκBα affects about 10% of all PRC2 target genes in the different models studied, and involves genes related with development, stemness and tissue homeostasis [4][5][7][13,14,16]. Importantly, IκBα deficiency leads to defective maturation of tissue stem cells, which are then retained in a fetal state [4][5][13,14]. The nuclear function of IκBα was first described in mammalian cells and tissues including skin [4][13] and intestine [5][14], but it is already present in organisms like Drosophila melanogaster [4][13] and Caenorhabditis elegans [7][16]. Notably, Caenorhabditis elegans , as the rest of nematodes, lacks recognizable NF-κB factors, strongly suggesting that nuclear and polycomb-related IκBα functions appeared in the evolution before or in parallel to its role as NF-κB inhibitor.
Therefore, there is cumulative data indicating that alterations related with IκBα function not only affect NF-κB pathway but IκBα exerts moonlighting functions including regulation of PRC2 activity on specific gene sets, which are pivotal for cancer initiation and progression.
2. IκBα in Hematologic Diseases
The first mutations of
NFKBIA, the gene codifying for IκBα, in cancer were detected in
Hodgkin lymphoma (HL), a B-cell lymphoma developed in lymph nodes, characterized by the presence of giant Reed-Sternberg (RS) cells. Several inactivating mutations (e.g., small insertions, deletions or nonsense mutations) in this gene are the direct cause of constitutive activation of the NF-κB pathway observed in RS cells. These mutations produce non-functional proteins, which usually lack fragments of the ankyrin domain and/or the C-terminal region. About 37% of HL patients show
NFKBIA mutations, with more than 18% of them having loss-of-function mutations (about 10% in both alleles)
[8][9][10][11][12][17,18,19,20,21].
Mutations in
NFKBIA were also found in around 22% of patients with
Gray zone lymphoma (GZL), a rare hematological disease with features intermediate between large B-cell lymphoma (LBCL) and classical HL. These mutations (non-sense or frameshift) are among the most prevalent in GZL patients
[13][22]A high presence of
NFKBIA mutations has also been reported in
diffuse large B-cell lymphoma (DCLBL) patients
[14][23] and in the RC-K8 DCLBL cell line
[15][24], which result in the total absence of the protein. Additional alterations, such as gene fusions involving
RELA have been found to impose constitutive NF-κB activation in these cells
[15][16][24,25]. Notably, high levels of NF-κB signaling involving nuclear c-Rel have also been observed in
mediastinal large B-cell Lymphoma (MLBCL), a subtype of DCLBL with clinical and molecular features that resemble the ones observed in HL patients
[17][26], which lack mutations in the coding region of NFKBIA
[18][27].
In addition to its linkage with B-cell neoplasms, aberrant IκBα function was also detected in
chronic myeloid leukemia (CML), a myeloproliferative disorder driven by the BCR-ABL translocation. In this type of cancer, IκBα is expressed at high levels and associated with the BCR-ABL and p53 proteins. This complex imposes cytoplasmic retention of p53 thus precluding its nuclear activity as tumor-suppressor
[19][28]. A crosstalk between IκBα and NF-κB with the tumor-suppressor p53 has also recently been identified and characterized in other oncogenic alterations such as tissue-specific lymphomas arising in the
Trp53-deficient mouse model
[20][29] and acute lymphoblastic leukemia cells
[21][30].
In
ectodermal dysplasia, mutations leading to N-terminal truncation of IκBα leading to a non-degradable protein with higher inhibitory activity on NF-κB have been identified
[22][31] but the mechanisms linking this particular alteration with disease remains unexplored.
Moreover, different hematological diseases have been linked to the presence of polymorphisms in the
NFKBIA gene likely associated with altered protein activity or gene expression. For example, polymorphisms localized in regions that may affect the rate of protein degradation are more frequent in
multiple myeloma patients than in healthy subjects
[23][24][32,33]. Some of these polymorphisms are also found in lymphoma
[9][10][18,19], further supporting their functional relevance.
In general, whether NFKBIA mutations promote cancer initiation and/or progression through activation of NF-κB signaling or by alternative NF-κB-independent mechanisms in the different systems has not directly been analyzed.
3. IκBα Loss in Glioma
It has consistently been observed that NF-κB activity is significantly higher in brain tumors than in normal tissues
[25][26][34,35]. Notably, analysis of a large set of
grade IV glioblastoma patients demonstrated reduced expression of IκBα in these tumors. Reduced IκBα levels in glioblastoma was not linked to mutations in the coding or promoter regions of the
NFKBIA gene, but to gene copy number alterations. Monoallelic deletions were detected at low frequency in classical glioblastoma (~6%) and were more common in non-classical subtypes (~25%). Notably, heterozygous
NFKBIA deletion and
EGFR amplifications are mutually exclusive suggesting that both alterations impact in the same pathway, which should be further investigated.
NFKBIA deletions and reduced IκBα levels are significantly associated to unfavorable outcomes and tumor recurrence in patients
[27][28][29][36,37,38]. In vitro experiments using glioblastoma cell lines demonstrated that the reintroduction of
NFKBIA gene or IκBα protein by nanoparticles induced apoptosis and increased the sensitivity of tumor cells to chemotherapy
[30][39]. In addition to allelic loses, a single-nucleotide polymorphism in exon 1
of NFKBIA, which leads to reduced expression of IκBα, is more frequently present in glioblastoma patients than in the healthy population and is associated to poor prognosis
[31][40].
In
lower-grade glioma (LGGs), which is a heterogeneous group of brain tumors that can progressively evolve into higher-grade gliomas, heterozygous
NFKBIA deletions are observed in approximately 7% of patients, being more common in the more advanced stage III tumors (~11%) than in stage II tumors (~3%). Moreover, it was demonstrated that both NFKBIA deletion and reduced levels of IκBα are associated with tumor aggressiveness and poor prognosis. In fact, the dosage of
NFKBIA was identified as an independent factor for 5-year survival and recurrence-free survival
[28][37].
4. Targeting NF-κB or/and Chromatin Editing Enzymes for Treating IκBα-Deficient Tumors
NF-κB has been considered as a targetable pathway in cancer for many years. However, general inhibition of NF-κB signaling was found to be extremely toxic due to its widespread effects (reviewed in
[32][79]). One of the first inhibitors targeting NF-κB that was approved by its use in multiple myeloma was Bortezomib, a reversible proteasome inhibitor that prevents IκBα degradation
[33][80]. Later on, it was shown that general NF-κB inhibitors led to massive inflammation in mice by increasing IL1-β secretion and inflammasome activation
[34][81], thus questioning the suitability of these inhibitors for cancer therapy. One possibility, which should be tested, would involve using short-duration treatments or treatments involving suboptimal NF-κB inhibitors dosage. Alternatively, specific tumors carrying defective IκBα activity but normal NF-κB activity may display chromatin alterations (due to defective moonlighting IκBα functions) that could be therapeutically targeted with PRC2 inhibitors that are currently under investigation
[35][82]. Further research on IκBα-deficient tumors will provide valuable information on novel druggable targets, other than NF-κB, that will benefit treatment of a specific group of cancer patients.