 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Muhammad Umer Ashraf | + 3149 word(s) | 3149 | 2021-09-24 10:59:04 | | | |
| 2 | Catherine Yang | -3 word(s) | 3146 | 2021-10-08 08:01:28 | | |
Video Upload Options
Epitranscriptomics has contributed greatly to the clinico-biological practices due to its diverse role in regulating at the post-transcriptional and translational levels. Epitranscriptomics is generally referred to chemical modifications in the RNA molecule without changing the nucleotide sequence. So far, more than 160 chemical modifications have been identified; playing a crucial role in regulating various biological processes, for example, in acute myeloid leukemia treatment, lung adenocarcinoma, gastric cancer and broad range tumor types.
1. Introduction
The advent of immunotherapy given in combination with standard chemotherapeutic drugs has greatly controlled cancer spread over decades. However, still, some cancer patients develop resistance against these therapeutic approaches, alarming the discovery of further advanced medicines. The invention of programmed cell death protein-1 and its ligand-1 (PD-1/PD-L1) was a breakthrough in the history of cancer treatment, but still, some tumors escape these immune surveillance mechanisms and relapse to grow continuously. Therefore, a more creative and advanced treatment is required instantly to overcome the issues largely associated with high-dose antibody/drug toxicities and drug resistances, conceivably in the form of personalized medicines. In this study, we have summarized epitranscriptomic mechanisms to improve the efficacy of ICB-therapy by targeting N 6A-modification machineries especially m 6A-modifiers. Moreover, we have also emphasized co-targeting immune checkpoint proteins (PD-1 and PD-L1) along with intracellular checkpoint molecules (CISH, SOCS-1 and microRNAs) in enhancing the efficacy of ICB-therapeutics by combining immunotherapy. A recent human clinical trial NCT04426669, NCT03538613 [1][2] evidenced the success of targeting CISH/SOCS-1 in NK-cells [3][4], T-cells [5][6], and DCs [7] in further strengthening the efficacy of ICB-therapy against a broad range of solid tumors and metastatic gastrointestinal cancers ( Figure 1 ).
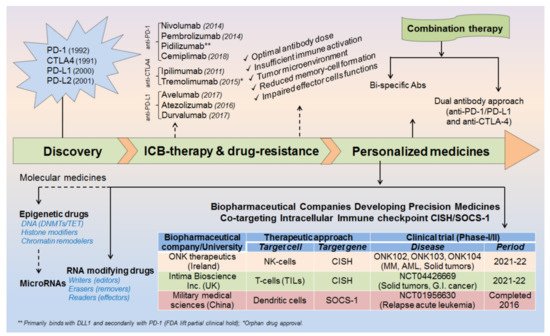
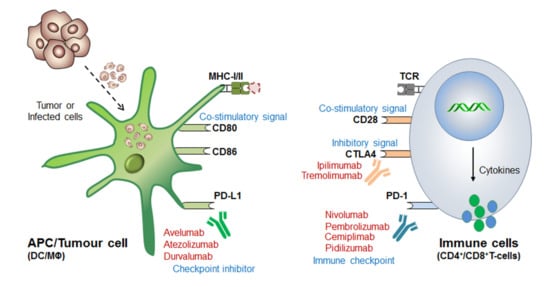
Before coming to the mainstream, a very logical question arises: (i) why even after so strong therapeutic approaches still some cancer cells escape these immune surveillance mechanisms? (ii) What could be the best possible combinations to overcome the issues associated with drug resistance and high-dose antibody toxicities [8][9][10][11] and (iii) what would be the best diagnostic biomarkers or alternative strategies to completely eliminate these cancerous cells? Such questions provoked the scientist to further understand in-depth the molecular mechanism of immune cell regulation and ICB-drug resistance. This revealed that; immune cells contain both inhibitory (break) as well as activator (acceleratory) markers to maintain immune homeostasis or to avoid a situation called autoimmunity and self-tolerance phenomena. This understanding led to the discovery of (i) first immune checkpoint marker PD-1 or PDCD1 (CD279) in 1992 [12] and (ii) immune cell inhibitory marker cytotoxic T-lymphocyte-associated protein-4 (CTLA-4 or CD152) in 1991 [13] or 1995 [14][15]. However the first anti-CTLA4-based therapy ‘Ipilimumab’ was approved in 2011 by (James P. Allison, Nobel laureate, physiology or medicine, 2018) Medarex and Bristol-Myers Squibb for the treatment of melanoma, and the first anti-PD-1 therapy was approved in 2014 for melanoma and in 2015 for non-small-cell lung carcinoma (NSCLC) treatment. Later, the tumor-cell inhibitory marker PD-L1 (CD274, previously known as B7-H1) was discovered in 1999-2000 [16] and PD-L2 (CD273, previously known as B7-DC) in 2001 [17] and was considered even much better control over immune cell checkpoint-based therapeutic targets [18] ( Figure 2 ).
Besides the patient‘s age, cancer stage (I–IV) and various environmental factors; there might be several other factors for increased drug resistance and reduced efficacy of ICB therapeutics [11]. For example, sub-optimal antibody dose, insufficient immune cell activation, intra-tumoral microenvironment, reduced memory cell formation and impaired effector cell functions after the first course of treatment schedule. Sometimes, high-dose antibody toxicity also becomes a major concern for its adverse consequences ( Figure 1 ). Therefore, a more advanced and unique therapy is required promptly to overcome these major issues.
Conclusively, our study devotes to improve the efficacy of ICB-therapy by co-targeting (i) epitranscriptomics (ii) intracellular immune checkpoints and (iii) microRNAs. More importantly, our investigation would help to design a specialized approach or custom-made strategies to improve the efficacy of ICB therapy [7][20][21].
2. Epitranscriptomics in ICB-Therapeutics
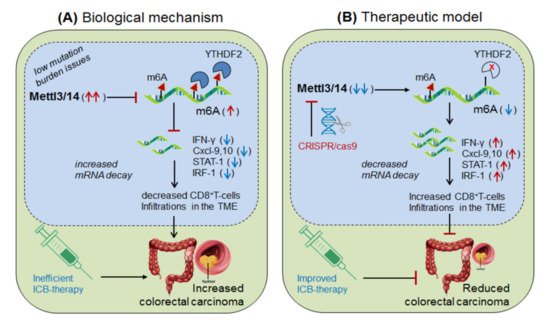
Wang, et al., 2020 [22] demonstrated the role of Mettl-3/14 (m6A-writer enzyme) in improving the efficacy of anti-PD-1 therapy. They found that even after standard anti-PD-1 treatment, still, some patients with colorectal cancer and melanoma develop resistance, because of insufficient immune response generated by the tumors with low mutation burden issues (mismatch-repair-proficient or microsatellite instability-low ‘pMMR-MSI-L’) constituting ~85% of the patients [23]. They found that these patients have significantly increased levels of Mettl-3/14, which has impaired the function of certain crucial genes under the tumor microenvironment (TME). Interestingly, CRISPR/cas9-mediated deletion of Mettl-3/14 in a colorectal cancer cell line (CT26) and murine melanoma cell line (B16) has not only increased cytotoxic CD8 + T-cell (CTL) infiltrations in the TME but also provided a durable adaptive immune response. Mechanistically, they justified that, the loss of Mettl-3/14 augmented mRNA-stability of IFNγ, STAT-1 and IRF-1 by promoting IFNγ-STAT1-IRF1-signalling through YTHDF2 reader proteins [24][25], leading to prolong secretion of these cytokines in the TME, resulting in the strong immune response. These investigations suggest the key role of Mettl-3/14 in inhibiting the efficacy of anti-PD-1 therapy by decreasing IFNγ, Cxcl-9 and Cxcl10-mediated immune response. Conclusively, this study endorsed the immunotherapeutic potential of m6A-writer in improving the efficacy of anti-PD-1 antibody by silencing Mettl-3/14 in the TME [22]. Moreover, overexpressing FTO (m6A-demethylase) or by targeting intracellular YTHDF2 (m6A-reader protein) in decreasing Mettl-3/14 methylation could be considered as an alternative strategy to improve anti-PD-1 therapeutics ( Figure 3 ).
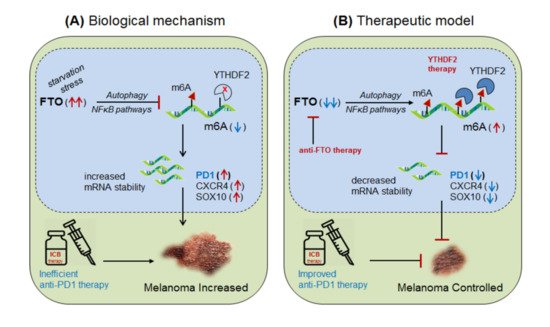
Yang et al., 2019 [26] demonstrated the role of m6A-eraser protein ‘FTO’ in melanoma progression, a type of skin cancer, and enlightened the intrinsic mechanism to improve the efficacy of anti-PD-1 therapy by targeting FTO. Yang and colleagues found that FTO is significantly up-regulated in human melanoma patients (metastatic skin samples n = 65) including human (Mel624) and mouse (B16F10) cell lines, and facilitated rapid tumorigenesis, caused by metabolic starvation stress in mice requiring autophagy and NFκB pathway [27]. However, selective depletion of ‘FTO’ not only increases sensitivity to anti-PD-1 therapy but also increases m6A methylation-inhibition of critical pro-tumorigenic (tumor-promoting) genes. Mechanistically, they proved that FTO-deficiency increases m6A-methylation at 5′UTR and 3′UTR of target genes; PD-1 (PDCD1), CXCR4 and SOX10, and thereby causing rapid mRNA-degradation by recruiting YTHDF2-reader proteins [28][24][25], confirmed by YTHDF2-knockdown in ‘increasing’ and YTHDF2-overexpression in ‘decreasing’ melanoma growth. Moreover, FTO-deficiency enhances the sensitivity of anti-PD-1 treatment by IFNγ-mediated cytokine response. These results clearly suggest that FTO plays a crucial role in melanoma tumorigenesis by regulating mTOR signalling through limiting the nutrient supply to the tumours [27]. Therefore, co-targeting FTO in combination with ICB-antibodies would be a promising approach to control melanoma progression [26]. This hypothesis was also supported by Singh et al., 2016 in controlling triple-negative inflammatory breast cancer cells using FTO (MO-I-500) inhibitor [29][30][31]. Theoretically, targeted overexpression of Mettl-3 might also control melanoma progression by decreasing FTO via balancing mechanism, and also by directly inhibiting the expression of pro-tumorigenic genes via recruiting YTHDF2 reader proteins ( Figure 4).
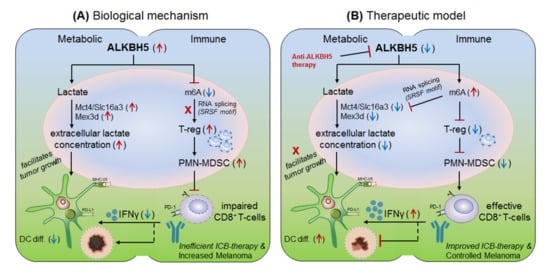
Li et al., 2020 [32] explained the role of another m6A-eraser protein ‘ALKBH5’ in the progression of melanoma-associated metastatic cancer, and enlightened the molecular mechanism to overcome anti-PD-1 resistance by targeting ALKBH5. Based on their previous studies [22] for the role of Mettl-3/14 in melanoma progression, the authors hypothesized that ALKBH5 might also have a significant role in regulating the efficacy of anti-PD-1 therapeutics. To this end, Li and colleagues used B16 (mouse melanoma) and CT26 (colorectal carcinoma)-induced TBM model, and selectively depleted ALKBH5 and/or FTO (CRISPR/Cas9-mediated silencing) in B16 and CT26 cell lines respectively, and injected subcutaneously into wild-type C57BL/6 and BALB/c mice to create tumor, followed by 1-day prior vaccination with irradiated B16 cells secreting GM-CSF ‘GVAX’ to induce sufficient antitumor T-cell response, and finally, anti-PD-1 antibody treatment was given to check its efficacy. Interestingly, ALKBH5 −/− TBM showed prolonged survival and slower tumor growth as compared to the non-transfected (NTC) control mice, suggesting the direct involvement of ‘ALKBH5’ in interfering with the efficacy of anti-PD-1 antibody. To further elucidate the role of ALKBH5 in modulating GVAX/anti-PD-1 treatment, they analysed tumor infiltrating lymphocytes (TILs) by FACS and found that among total CD45 + CD4 + CD8 + gated populations, ALKBH5 −/− mice have elevated granzyme-B (GZMB) + CD8 + , GZMB + CD4 + T-cell, NK-cell (CD56 + ) and dendritic cell (DCs: CD45 + Ly6C - MHC-II + CD24 hi F4/80 lo ) numbers, but more importantly, T-reg (CD4 + Foxp3 + ) and polymorphonuclear myeloid-derived suppressor cell (PMN-MDSCs: CD45 + CD11b + Ly6G + Ly6C lo F4/80 − MHC-II − ) populations were drastically reduced as compared to the control mice. This was further validated by immunohistochemistry staining (IHC) of the MDSC-mLy6G, however, no differences in other immune cell populations (MDSC and macrophage) were noted. This suggests that ALKBH5 has the potential to recruit immunosuppressive (T-reg and PMN-MDSCs) populations in the TME during ICB therapy. Again, to stamp the selective function of immunosuppressive cells in inhibiting the anti-PD-1 effect, they specifically depleted T-regs (anti-CD25) and PMN-MDSCs cells in the NTC control mice, resulting in delayed tumor progression as compared to the ALKBH5 −/− model (due to already fewer T-reg numbers), confirming the immunosuppressive function of T-regs (induced by MDSCs) in impairing the efficacy of anti-PD-1 antibody by inhibiting CD8 + T-cells effector functions through decreasing DC-differentiation (CD45 + Ly6C - MHC-II + CD24 hi F4/80 lo ) markers [33]. These observations clearly suggest that ALKBH5 recruits immunosuppressive populations in the TME and thereby interfering with the efficacy of anti-PD-1 therapy. Next, to identify the molecular targets, they sequenced RNA isolated from ALKBH5/FTO −/− B16 tumors and compared it with the NTC-control TBM on day-12 after GVAX/anti-PD1 treatment. Interestingly, the gene ontology (GO) analysis of the differentially expressed genes (DEG) revealed ALKBH5 is associated with metabolic genes especially ‘Mct4/Slc16a3’ involved in lactate metabolism, whereas, FTO is associated with IFNγ and chemokine signalling pathways. This was validated by increased IFNγ intermediates (qRT-PCR expression) upon in-vitro stimulation of IFNγ to the FTO −/− B16 cells. Moreover, the comparison of mouse DEGs with human melanoma patients (n = 21 anti-PD1 therapy responders) and (n = 17 non-responder) reveals eight common genes associated with ALKBH5-deficiency and eleven common genes with FTO-deficiency, indicating ‘conserved’ and potential targets of ALKBH5 and FTO in mouse as well as human receiving anti-PD1 therapy. This suggests that ALKBH5 modulates anti-PD-1 resistance by recruiting immunosuppressive T-reg cells and by modulating metabolic genes whereas FTO works by targeting IFNγ and by modulating inflammatory chemokine-mediated signalling pathways in the TME. Next, epigenetic analysis via LC-MS/MS reveals higher m6A-abundance in ALKBH5-deficient as compared to FTO-deficient B16 tumours, which meaningfully suppresses the expression of m6A-mediated ‘Mct4/Slc16a3’ in ALKBH5 alone and ‘Mex3d’ in ALKBH5 and FTO both. Moreover, MeRIP-seq reveals enriched SRSF motif (a subunit of SAG core involved in RNA splicing [34]) in ALKBH5-deficient tumors as compared to FTO, suggesting different mechanisms of action of these two de-methylases in modulating anti-PD1 efficacies. Collectively, these results suggest that ALKBH5 and FTO target metabolic genes and increase the expression of Mct4/Slc16a3 and Mex3d (supplementing lactate to the tumour) by inhibiting m6A methylation-mediated RNA-splicing mechanisms, supported by Zaho et al., 2014 [35], ( Figure 5, therapeutic model). Furthermore, to dig out the m6A-modulated genes via RNA-splicing mechanism, they identified m6A-enriched transcripts around 5′-3′ splice sites by m6-CLIP and found the involvement of three immunotherapeutic resistance genes Eif4a2, Arid4b and USP15 affecting the response of anti-PD-1 therapeutics by regulating transcription, translation and T-reg activation via TGF-β signalling in the TME. ( Figure 5) Taken together, ALKBH5 is playing a crucial role in promoting tumour metastasis, and therefore intracellular silencing of ALKBH5 in the TME would hold the potential to control tumor metastasis via increasing the efficacy of anti-PD-1 therapeutics [32].
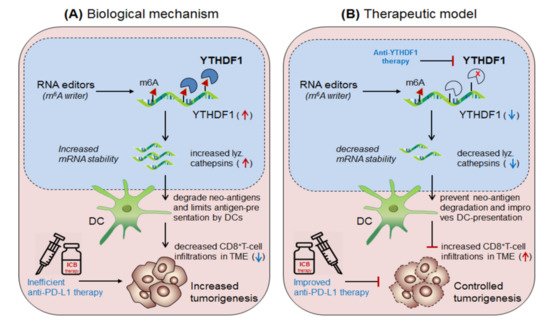
Han et al., 2019 [37] demonstrated the synergistic role of dendritic cells expressing ‘m6A-writer’ and ‘YTHDF1-readers’ proteins in anti-tumor immunity. They found that despite the presence of numerous neo-antigens, some patients still failed to generate sufficient anti-tumor response. To this end, in discovering the intrinsic molecular mechanism, they generated dendritic cell-specific conditional knockout mice depleted with YTHDF1 (YTHDF1 cKO ) gene. Surprisingly, the loss of YTHDF1 enhances antigen-recognition and cross-presentation ability of DCs in-vivo, resulting in elevated CD8 + T-cell infiltration in the TME as compared to the control wild type (YTHDF1 WT ) mice. Moreover, YTHDF1 cKO mice showed an enhanced response to anti-PD1 therapy [23]. Mechanistically, they proved that the wild type mice, in the presence of m 6A mRNA-methylation machineries recruited YTHDF1 reader proteins at the lysosomal-cathepsins mRNA axis, resulting in increased mRNA-stability, and thereby increased the abundance of cathepsin proteins in the phagosomal compartments of the DCs, causing severe degradation of the neo-antigens and thus limiting the antigen availability to the DCs for antigen-recognition and further cross-presentation to CD8 + T-cells in the cytosol. This result suggests that YTHDF1 is playing a crucial role in suppressing anti-tumor immunity [37], and therefore intracellular silencing of YTHDF1 in DCs designates its potential to enhance anti-tumor immunity. Collectively, this discovery reveals two important mechanisms to enhance anti-tumor immunity by co-targeting (i) anti-YTHDF1 therapy: where, YTHDF1-deficiency protect ‘antigen degradation' and allows efficient recognition and presentation by DCs, in-turn, further increases the abundance of DC-mediated effector CD8 + T-cells by cross-presentation mechanism, supported by Ding et al., 2021 [38] and (ii) by enhancing anti-PD-1/PD-L1 efficacy: which further potentiates the efficacy of anti-PD-1 immunotherapy by enhancing the effector function of CD8 + T-cells in the TME [23] Figure 6.
References
- Palmer, D.; Webber, B.; Patel, Y.; Johnson, M.; Kariya, C.; Lahr, W.; Parkhurst, M.; Gartner, J.; Prickett, T.; Lowery, F.; et al. 333 Targeting the apical intracellular checkpoint CISH unleashes T cell neoantigen reactivity and effector program. J. Immunol. Ther. Cancer 2020, 8, A204.
- Plieth, J. Crispr: Nice Valuation, but Where’s the Clinical Trial? Available online: https://www.evaluate.com/node/13152/pdf (accessed on 20 July 2021).
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824.
- Putz, E.M.; Guillerey, C.; Kos, K.; Stannard, K.; Miles, K.; Delconte, R.B.; Takeda, K.; Nicholson, S.E.; Huntington, N.D.; Smyth, M.J. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology 2017, 6, e1267892.
- Palmer, D.C.; Webber, B.R.; Patel, Y.; Johnson, M.J.; Kariya, C.M.; Lahr, W.S.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Lowery, F.J.; et al. Internal checkpoint regulates T cellneoantigen reactivity and susceptibility to PD1 blockade. bioRxiv 2020.
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113.
- Wang, D.; Huang, X.F.; Hong, B.; Song, X.T.; Hu, L.; Jiang, M.; Zhang, B.; Ning, H.; Li, Y.; Xu, C.; et al. Efficacy of intracellular immune checkpoint-silenced DC vaccine. JCI Insight 2018, 3.
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities Associated With PD-1/PD-L1 Blockade. Cancer J. 2018, 24, 36–40.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16.
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164.
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39.
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895.
- Bashyam, H. CTLA-4: From conflict to clinic. J. Exp. Med. 2007, 204, 1243.
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988.
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547.
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369.
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268.
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472.
- Zhao, X.; Yuan, C.; Wangmo, D.; Subramanian, S. Tumor-Secreted Extracellular Vesicles Regulate T-Cell Costimulation and Can Be Manipulated To Induce Tumor-Specific T-Cell Responses. Gastroenterology 2021, 161, 560–574.
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61.
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68.
- Wang, L.; Hui, H.; Agrawal, K.; Kang, Y.; Li, N.; Tang, R.; Yuan, J.; Rana, T.M. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020, 39, e104514.
- Kim, D.J.; Iwasaki, A. YTHDF1 Control of Dendritic Cell Cross-Priming as a Possible Target of Cancer Immunotherapy. Biochemistry 2019, 58, 1945–1946.
- Hou, G.; Zhao, X.; Li, L.; Yang, Q.; Liu, X.; Huang, C.; Lu, R.; Chen, R.; Wang, Y.; Jiang, B.; et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Res. 2021, 49, 2859–2877.
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120.
- Yang, S.; Wei, J.; Cui, Y.H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782.
- Gulati, P.; Cheung, M.K.; Antrobus, R.; Church, C.D.; Harding, H.P.; Tung, Y.C.; Rimmington, D.; Ma, M.; Ron, D.; Lehner, P.J.; et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc. Natl. Acad. Sci. USA 2013, 110, 2557–2562.
- Dai, X.Y.; Shi, L.; Li, Z.; Yang, H.Y.; Wei, J.F.; Ding, Q. Main N6-Methyladenosine Readers: YTH Family Proteins in Cancers. Front. Oncol. 2021, 11, 635329.
- Kumar, S.; Nagpal, R.; Kumar, A.; Ashraf, M.U.; Bae, Y.S. Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines 2021, 9, 690.
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.; Olsen, M.; Lucci, A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS ONE 2016, 11, e0159072.
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.
- Li, N.; Kang, Y.; Wang, L.; Huff, S.; Tang, R.; Hui, H.; Agrawal, K.; Gonzalez, G.M.; Wang, Y.; Patel, S.P.; et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 20159–20170.
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434.
- Adhikari, S.; Xiao, W.; Zhao, Y.L.; Yang, Y.G. m(6)A: Signaling for mRNA splicing. RNA Biol. 2016, 13, 756–759.
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419.
- Selberg, S.; Seli, N.; Kankuri, E.; Karelson, M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6, 13310–13320.
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274.
- Ding, Z.; Li, Q.; Zhang, R.; Xie, L.; Shu, Y.; Gao, S.; Wang, P.; Su, X.; Qin, Y.; Wang, Y.; et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal. Transduct. Target. Ther. 2021, 6, 26.


