1. Increased fragility fractures in T2DM patients with normal to higher bone mineral density: BMD does not predict bone fragility in this metabolic phenotype.
Increased fragility fractures are well documented in patients with type 2 diabetes mellitus (T2DM), a condition of chronic hyperinsulinaemia [1][2][3][4]. Decreased skeletal bone mineral density (L-BMD) is the phenotype of “classical” osteoporosis [5][6]. A higher BMD is considered to confer greater bone strength (fracture resistance). However, the T2DM bone fragility phenotype more often presents with normal to increased BMD (H-BMD) [4][6][7][8][9][10][11], and positively tracks with increased fracture risk [1][2][4][9][12].
In a cross-sectional non-intervention study of 146 Caucasian non-diabetic postmenopausal women with a mean age of 60 ± 2.7 years, HOMA-IR was found to be positively associated with volumetric bone mineral density (vBMD) [8]. In addition, increased insulin resistance (IR) and treatment with exogenous insulin therapy is positively associated with higher BMD and increased fragility fractures. A study following 5994 T2DM males ≥ 65 years of age, found a higher non-vertebral fracture risk in those using insulin (HR 1.74, 95% CI 1.13, 2.69) who also had a higher BMD [12]. Furthermore, in a population-based matched cohort study investigating primary care records of 2979 insulin users and 14,895 non-users, T2DM patients exposed to insulin therapy to manage glycaemia were found to have a 38% excess fracture risk [9]. In both H-BMD-associated T2DM “osteofragilitas” (bone fragility) and L-BMD osteoporosis phenotypes, there is an increase in bone fragility and a loss in tensile and/or torsion strength, and bone ductility, resulting in higher rates of fractures.
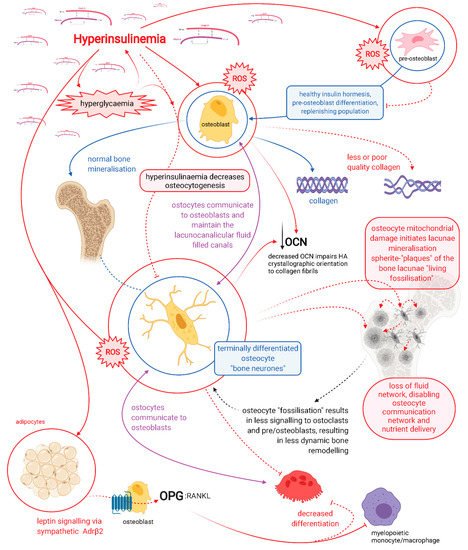
Hyperinsulinaemia drives the pathogenesis of T2DM, which may precede hyperglycaemia by up to 24 years [3][13]. Hyperinsulinaemia decreases osteoblastogenesis and propagates poorer-quality collagen production, a problem further compounded by hyperglycaemia increasing glycation damage on new or existing bone collagen. Hyperinsulinaemia drives chronic osteocyte distress via excess ceramide synthesis, which increases cellular reactive oxygen species (ROS) [14][15][16][17], leading to a unique type of mineralisation within their lacunae, coined by Bell, Kayser and Jones as “living fossilisation” [18][19][20][21][22]. Concomitantly, hyperinsulinaemia decreases osteoclastogenesis, thus impeding the bone resorption needed to enable homeodynamic bone remodelling—a marker of good health [4][23] —and this results in a form of hyperinsulinaemia-hyperglycaemia pseudo-osteopetrosis. Combined, these hyperinsulinaemia-driven effects result in the increased bone mineral density seen in people with T2DM.
Osteocytes are the backbone of bone health, and consequently major players in whole-body metabolism [24]. Osteocytes are able to directly mineralise and resorb bone and are central mediators in the regulatory control of osteoblasts and osteoclasts [25][26][27][28][29][30][31][32][33]. Chronic hyperinsulinaemia diminishes the replenishment of osteocytes and drives the living fossilisation of the ones in existence, leading to the loss of the osteocyte’s dynamic orchestration of bone remodelling [18][19][20][21][22][34]. We propose chronic hyperinsulinaemia provides a plausible explanation, a unifying theory of the mechanisms of action, for the increased BMD and bone fragility “osteofragilitas”, that leads to the increased fracture rates seen in the T2DM bone phenotype.
2. Osteocytes: Mediators of Bone Remodelling and Metabolic Heath
Within the adult skeleton, osteocytes comprise 90% to 95% of the total bone cells [34]. Osteocytes embed within the bone after collagen formation by osteoblasts, of which some of these osteoblasts are fated to differentiate into the embedding osteocytes [34]. The osteocytes, along with their neighbouring osteoblasts, continue to form mineralised bone onto the collagen scaffold, in the process forming the hydroxyapatite lacuna chamber around the embedding osteocytes [24]. Continual morphological changes take place in the process of osteocytogenesis, leading to cells that bear little resemblance in structure to their predecessors, appearing visually more like neurons. Osteocytes have on average 50, and up to 100 dendrites, extending through highly connected intricate tunnels, called the canaliculi. The canaliculi are formed and maintained by the occupying osteocytes [35], and enable physical connection to other osteocytes and to the outside surface of bone, to osteoblasts, osteoclasts and the vasculature [36].
The lacunocanalicular system is a fluid-filled space, separated from the mineralised component of bone and maintained by the resident osteocytes [19]. The osteocytes mediate bone remodelling, sense and respond to mechanical stress via their strategic distribution and network of a vast number of connected dendritic processes that enable intercellular communication. Their network detects fluid shear stress, enabling the translation of mechanical stimuli into biochemical messages. Osteocytes feed these signals forward via autocrine and paracrine mechanisms, to elicit homeostatic adaptive responses, which include: regulating bone remodelling in order to provide sensitive and continual changes to whole-body mineral needs, to effecting distant organ responses via endocrine signalling[19][36].
3. Dendritic Connectivity Is Essential for Function and Viability
Osteocytes connect to one another via their dendritic processes which form the osteocyte-lacunocanalicular network, and their health and viability are dictated by their dendritic connectivity [36][37]. A compromise in osteocyte health diminishes their ability to actively inhibit mineralisation of their pericellular space [22][25], consequently reducing their connectivity. This further compromises the health of deeper osteocytes that become cut off from receiving nutrients which are no longer able to be delivered through fluid movement via the canalicular tunnels. Furthermore, osteocyte connectivity is required for the transduction of load-induced fluid flow that enables their mechanical sensory system to decrease apoptosis and increase osteocytogenesis [38]. Maintenance of dendritic connectivity allows osteocyte-directed regulation of osteoblasts and osteoclasts, in addition to their own capacity to directly resorb bone [26][27][31][32][33].
It is highly likely that osteocytes rather than osteoclasts are responsible for bone resorption in the basal condition, as a function of whole-body mineral homeostasis, and that osteoclast bone resorption serves to function in the acute action/need/stress response [31]. Patients with T2DM have decreased levels of carboxy-terminal collagen crosslinks (CTX) bone resorption marker, indicating a lower bone turnover [10]. Osteocytes regulate both osteoblast and osteoclast differentiation and function, and are thus master regulators of dynamic bone remodelling, a function of healthy physiology (Figure 1) [39].
Figure 1. Schematic representation showing the dynamic role of osteocytes in the regulation of healthy and dysregulated bone. Beta-adrenergic receptor (Adrβ2), hydroxyapatite (HA), osteocalcin (OCN), osteoprotegerin (OPG), receptor activator of nuclear factor kappa-β ligand (RANKL), and reactive oxygen species (ROS). Red lines indicate hyperinsulinaemia-driven pathology pathways. Healthy physiology indicated with blue and purple lines.
4. Hyperglycaemia Increases Advanced Glycation End-Product Formation in Bone Collagen
The anabolic hormone insulin is required at the basal level for healthy bone formation . However, chronic hyperinsulinaemia surpasses this threshold in a dose and duration manner. When coupled with hyperglycaemia, it results in the production of poorer-quality, more rigid collagen and glycation damage of existing collagen [40][41]. Together, they drive increasing BMD by promoting skeletal mineral acquisition that is fragile in structure [19][42]. Hyperglycaemia and hyperinsulinaemia increase advanced glycation end-product (AGE) formation. Increased glycation on fibrillar collagen negatively affects bone quality [11]. In human tissue, the most abundant AGE is glucosepane, a lysine–arginine cross-linking, that forms the major AGE in bone type 1 collagen. Hyperglycaemia is one of the leading causes of AGE formation, affecting the structural and biochemical properties of protein binding sites, rendering them unrecognisable to other proteins and enzymes [41].
Hyperglycaemia-driven AGE formation of glucosepane in bone collagen causes a decrease in viscoelasticity and increases the production of a stiffer collagen, resulting in negative effects on the mechanical properties of load-bearing collagen in bone. This causes bone toughness to decrease, while a greater accumulation of AGE in bone results in increased fracture risk [41]. Furthermore, an increase in stiffer/rigid collagen production occurs due to increased glycation effects on the vasculature, leading to increased hypoxia in the microenvironment [11]. Hypoxia then compromises the osteoblasts’ capacity to generate sufficient ATP for collagen synthesis and for differentiation into osteocytes (Figure 1). Both of which are very energy intensive processes, requiring an efficient mitochondrial capacity to generate ATP via oxidative phosphorylation (OxPhos) [43][44][45][46]. In addition, hyperinsulinaemia and hyperglycaemia inhibit beta-oxidation, whilst increasing mitochondrial (mt) reactive oxygen species (ROS) formation [47]. This leads to increased H2O2 production, causing damage to intracellular protein synthesis machinery and consequently synthesis of poorer quality collagen. The typical methodology of assessing bone quality is via dual x-ray absorptiometry, however, this method is unable to detect the collagen aspect of bone quality [11]. As a result, there is an increased frequency in missing early detection of hyperinsulinaemia-hyperglycaemia osteofragilitas fracture risk, which typically does not present with L-BMD. This suggests that BMD alone is a poor proxy/diagnostic marker for fracture risk in hyperinsulinaemic individuals.
Hyperinsulinaemia “enforces” cellular glucose substrate fuelling [48], and downregulates beta-oxidation by increasing intracellular ceramide production [17]. Excess ceramide production compromises mtOxPhos capacity, by increasing dynamin-related protein 1 (Drp1) synthesis. Drp1 functions to increase mitochondrial fission, in addition to increasing the production of mtROS such as: superoxide (O2−), hydroxyl radical (−OH) and hydrogen peroxide (H2O2) [14][17][49]. Concomitantly, ATP production from glucose oxidation reduces the intracellular pool of nicotinamide adenine dinucleotide (NAD+), consuming four NAD+ in the production of two acetyl moieties, in comparison to beta-oxidation, ketolysis or oxidation of acetoacetate, which consume two, one and zero respectively (Figure 2) [50]. Thus, ATP synthesis that is increasingly reliant on glucose oxidation, negatively impacts the availability of NAD+.
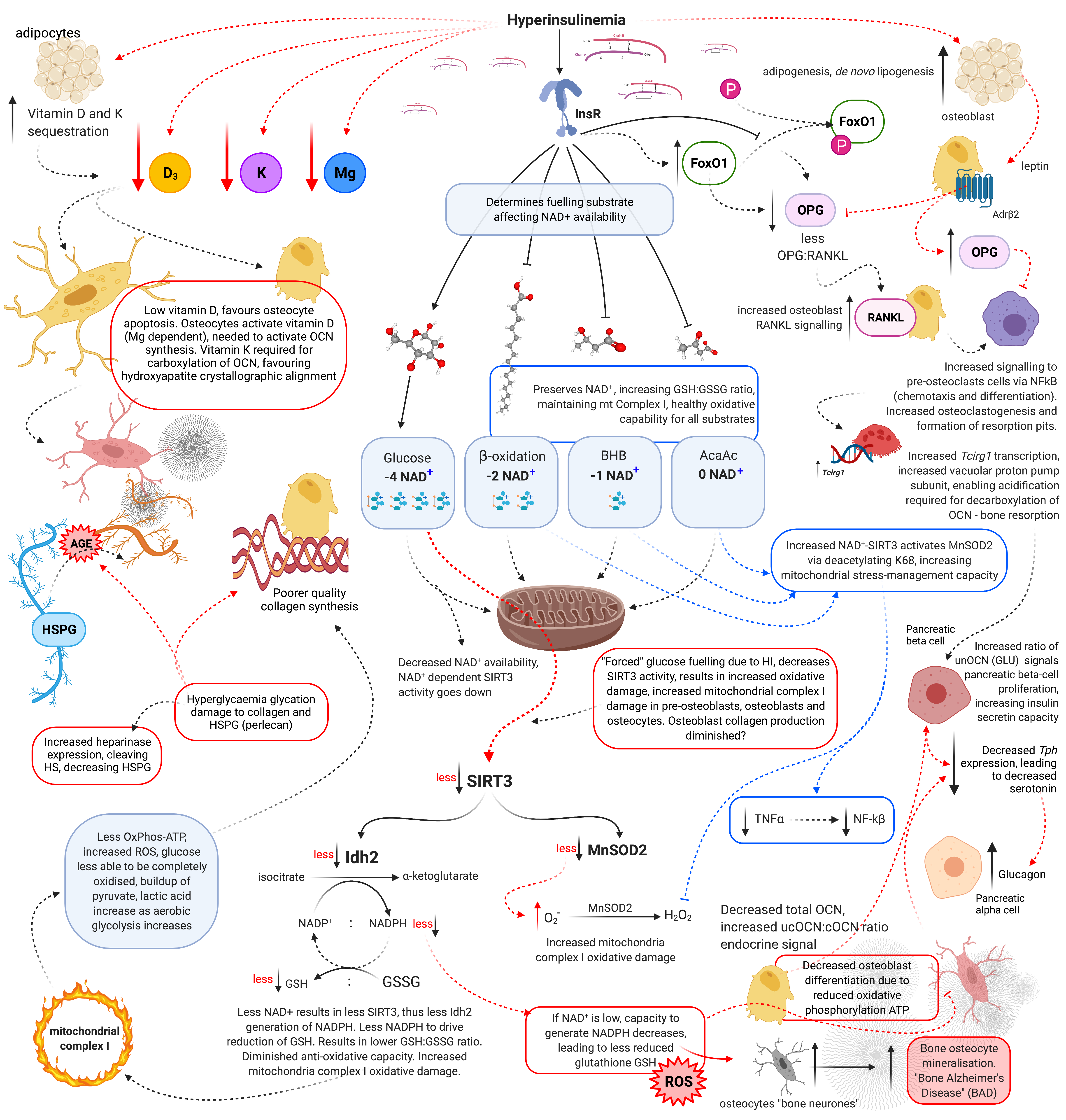
Figure 2. Schematic representation of hyperinsulinaemia effects on cellular oxidative state, and bone homeodynamics. Acetoacetate (AcAc), adenosine triphosphate (ATP), advanced glycation end-products (AGE), beta-adrenergic receptor 2 (AdrB2), beta-hydroxybutyrate (BHB), heparan sulphate (HS), heparan sulphate proteoglycan (HSPG), hydrogen peroxide (H2O2), hyperinsulinaemia (HI), insulin receptor (InsR), isocitrate dehydrogenase 2 (Idh2), forkhead box O1 (FoxO1), glutathione oxidised form (GSSG), glutathione reduced form (GSH), lysine 68 (K68), magnesium (Mg), manganese superoxide dismutase 2 (MnSOD2), nicotinamide adenine dinucleotide (NAD+), nicotinamide adenine dinucleotide phosphate (NADP+), nuclear factor-kB (NF-kB), osteocalcin carboxylated (Gla-OCN or cOCN), osteocalcin un(der)carboxylated (Glu-OCN or ucOCN), osteoprotegerin (OPG), oxidative phosphorylation (OxPhos), receptor activator of nuclear factor-kB ligand (RANKL), reactive oxygen species (ROS), sirtuin 3 (SIRT3), superoxide (O2−), tumour necrosis factor α (TNFα), T cell immune regulator 1 (Tcirg1), tryptophan hydroxylase (Tph), vitamin D (D3), and vitamin K (K).
Sirtuin-3 (SIRT3) regulates the synthesis of endogenous antioxidant enzymes such as mitochondrial manganese superoxide dismutase (MnSOD2) and NADPH-dependent production of reduced glutathione (GSH) [50][51]. An increased reliance on glucose fuelling and its effect on NAD+ availability, leads to a reduction in SIRT3 activity, since SIRT3 is NAD+ dependent. This in turn decreases signals for the transcription and synthesis of MnSOD2 and GSH [50][51]. Furthermore, hyperinsulinaemia, diminishes the oxidative buffering capacity of the cellular redox antioxidant GSH [52]. Beta-oxidation activity dramatically increases in osteoblasts as they mature. If beta-oxidation is diminished and/or inhibited, metabolic demand cannot be met, resulting in a decrease in precursor cellular differentiation capability, as osteoblastogenesis and osteocytogenesis are energy intensive processes [43][45][46]. Bone neither stores nor synthesises any significant amount of fat. Thus, fatty acids delivered via chylomicron remnants (CR) and low-density lipoproteins (LDL) to bone are more likely to be used for ATP synthesis via beta-oxidation [45].
5. Hyperinsulinaemia Increases Osteocyte Mitochondrial Fission and Disassociation from the Endoplasmic Reticulum
Healthy osteocytes are able to transfer their functional mitochondria to neighbouring distressed osteocytes via their physically connected dendrites
[37]. Mitochondrial transfer between osteocytes declines with age and with decreased dendritic connections
[19][25][37][53]. Osteocytic rescue of their distressed neighbours via mitochondrial transfer is dependent on their mitochondria associating with the endoplasmic reticulum (ER) and on their dendrite connectivity, which requires the maintenance of their lacunocanalicular tunnels via inhibiting excessive mineralisation
[18][26][28][37]. Hyperinsulinaemia increases mtROS production via ceramide synthesis which increases mitochondrial fission and ER stress
[17][54]. This results in less mitochondrial fusion, which is necessary for beta-oxidation, and also leads to decreasing the osteocytes’ capability to transfer mitochondria to adjacent distressed osteocytes. This osteocytic mitochondria transfer process is dependent on mitochondrial association with the ER (ER-mito), driven by the protein guanosine triphosphatase mitofusin 2 (Mfn2), which localises to the outer mitochondrial membrane (OMM), enabling the tethering of the mitochondrion to the ER
[37]. Reductions in Mfn2 expression leads to reductions in mitochondrial fusion and distribution, which further decreases the ability of healthier osteocytes to rescue their distressed neighbours
[19]. Hyperinsulinaemia increases the Drp1:Mfn2 ratio. As a result this favours mitochondrial fission and ER-mito disassociation, leading to decreased OxPhos ATP synthesis and increased mtROS production
[17][55]. Interestingly, osteocytes are under a form of social control, as they must receive signals from other cells for their survival, which is reliant on their connectivity
[42]. A fall in the number of viable osteocytes or dendritic connections, below an essential minimum number (a threshold), or excessive lacunocanalicular mineralisation, may result in a severely compromised osteocyte-network signal transduction capacity, which in turn would impair dynamic bone remodelling
[42]. Osteocytes actively maintain the lacunae-canalicular space, enabling fluid flow dynamics that ensures the provision of nutrients and signalling molecules such as growth factors and cytokines, in addition to facilitating the removal of waste. This is essential for the deeper embedded osteocytes and osteoblasts, to remain viable and function healthily
[26]. Chronic hyperinsulinaemia may contribute to significant detrimental remodelling of dendrites and the lacunae-canalicular space, where the number (threshold) of osteocytes affected may result in a profoundly negative outcome.
6. MnSOD2 and SIRT3 Required for Osteoblastogenesis and Osteocytogenesis
Chronic hyperinsulinaemia and hyperglycaemia drives the pathogenesis of T2DM, chronic kidney disease (CKD) and atherosclerosis, conditions that are associated with increased rates of fractures independent of L-BMD. Hyperinsulinaemia with hyperglycaemia likely causes a decrease in osteocyte population and/or functional capacity leading to hyperinsulinaemia-
osteofragilitas [1][56][57][58][59]. Glucose restriction increases osteoblast-to-osteocyte specification, and conversely hyperglycaemia reduces osteoblast gene expression of
Osx, Bglap or
Dkk1, leading to a reduction in osteocytogenesis
[34]. Furthermore, hyperglycaemia decreases osteocyte connectivity and population. Viable osteocytes are needed to signal pre-osteoblastogenesis and osteocytogenesis. Therefore, hyperglycaemia’s negative impact on osteocyte numbers and dendritic connectivity, may result in a detrimental impact on the maintenance of the osteocyte-lacunocanalicular network. As a result, this would impair the osteocytes’ role in dynamic bone remodelling in the basal state. This is likely a contributing factor to the lowered state of bone remodelling seen in hyperinsulinaemic T2DM and CVD patients
[39].
Hyperinsulinaemia/glycaemia mediated decreases in NAD+ availability, consequently decreases antioxidants MnSOD2 and SIRT3 activity, this leads to a negative impact on osteoblast differentiation
[60]. Both MnSOD2 and SIRT3 are required to regulate mitochondrial stress, to enable cellular differentiation and bone formation
[60]. MnSOD2 dismutes mitochondrial superoxide, thus protecting complexes I and II of the mitochondrial electron transport chain (ETC), where OxPhos occurs. Glucose restriction increases ketolysis, which increases the redox span between complex I and III
[48][61]. This decreases electron leakage at these sites, which reduces the formation of superoxide. Glucose restriction concomitantly leads to an increase in NAD+ dependent SIRT3 activity, which increases deacetylation of MnSOD2 lysine residues: 53, 68 and 89 (K53, K68, K89), consuming NAD+ in the process
[50][51][62][63][64][65]. This consequently upregulates MnSOD2 antioxidant activity, resulting in improved ROS management
[66]. With improved ROS management and mt-OxPhos capacity, due to glucose restriction and ketolysis, this maintains osteocytogenesis, viability and connectivity. Maintenance of osteocyte viability knock-on effect results in sustained dynamic bone remodelling, as well as regulation of osteoblast and osteoclast differentiation and activity.
7. Glucose Restriction Increases Glutathione Activity and Improved Cell Viability
Glucose restriction decreases insulin signalling, thereby enabling increased beta-oxidation and ketolysis, preserving NAD+ availability to enable upregulated SIRT3 activity which increases NADPH production
[67]. This drives increased glutathione reductase activity, which increases the ratio of reduced to oxidised glutathione (GSH:GSSG). Thus enhancing the intracellular antioxidant capacity to combat ROS damage (
Figure 2)
[68].
Calorie restriction decreases glucose and insulin levels, whilst increasing fatty acid oxidation and ketogenesis. This is the metabolic phenotype of fasting. Mice under lower levels of glucose and insulin show a marked increase in SIRT3 expression and activity, consequently preventing age-related hearing loss, a condition associated with ageing, increased hyperinsulinaemia, and osteofragilitas conditions
[69]. In a mouse model, elevated insulin and high glucose compromised mitochondria in osteocytes, increasing both cytoplasmic and mitochondrial ROS, driving an inverse correlation between glutathione and mitochondrial ROS. Additionally, an increasing compromise in osteocyte mitochondrial-function strongly correlates with increased impairments in skeletal health
[70].
8. Glucose Restriction Enables Osteocytogenesis; Hyperglycaemia Inhibits It
Hyperglycaemia inhibits osteoblast differentiation into osteocytes, and reduces the osteoblasts bone mineralisation capacity. In an in vitro model, IDG-SW3 cells differentiated into osteoblasts were cultured in 1, 5 or 25 mmol/L of glucose, using an alizarin red assay, it was shown that the osteoblasts cultured in the higher glucose concentration had a decreased ability to form calcium deposits
[34]. Furthermore, osteocytes cultured in 25 mmol/L of glucose, showed a decrease in osteoblast-to-osteocyte transition, assessed using osteocytic
Dmp-GFP gene expression reporter. A negative correlation was found between glucose availability and osteocytogenesis, where osteoblast-to-osteocyte transition increased as glucose concentration decreased, assessed via fluorescence microscopy/cell sorting of
Dmp-GFP expression. In short, glucose restriction increased osteocyte gene expression
[34]. These results indicate hyperglycaemia may disturb, if not diminish osteoblast and osteocyte replenishment potential. Hyperinsulinaemia and hyperglycaemia impair osteoblastogenesis, osteocytogenesis and osteoclastogenesis, resulting in decreased dynamic bone remodelling. Patients with T2DM and CVD show marked decrease in bone remodelling
[4][45].
In a situation of hyperinsulinaemia which potentiates and consolidates hyperglycaemia, osteoblast bone formation may become compromised over time, while osteoblast-to-osteocyte transition is also diminished. In this scenario, one would expect less evidence of bone mineralisation. However, if in the same hyperinsulinaemic and hyperglycaemic state, the existing osteocytes are also producing elevated levels of mtROS, osteocyte death would potentially incur vacancies in their lacunocanalicular space, which would result in a state of heightened structural fragility. This may phenotypically appear as L-BMD osteoporosis. However, hypothetically in a hyperinsulinaemic state, the “bone neurone” sensory osteocyte makes a “margin-call”
[22][71][72], to fossilize instead of leaving an empty and thus structurally fragile space, so as to ensure structural rigidity at the very least.
Alternatively, the rationale may be simpler, where the osteocyte fossilisation occurs in the hyperinsulinaemic state, so as to aid in mineral accretion during times recognised as “feasting and energy abundance”. A plausible explanation would be hyperinsulinaemia occurring in summer and autumn, when there is greater availability of high carbohydrate foods, leading to increased levels and duration of insulin signals to the cells that there is a food abundance period. Insulin mediates de novo adipogenesis, chronic insulin signals to adipocytes to increase storage of fuel in the form of lipids (de novo lipogenesis) and to bone cells to save minerals “for a rainy day”, in expectation of typically less food abundance in winter. A similar physiological adaptation example for increasing storage of fuel, minerals and vitamins, is during pregnancy, which has a natural state of mild hyperinsulinaemia, in-order to provide for post-partum lactation
[32][73].
Glucose restriction would historically normally occur during wintertime, with concomitant lowered insulin levels, as a result of decreased food abundance, especially the farinaceous kind. A low glucose and low insulin environment would facilitate bone remodelling and improved resorption, as beta-hydroxybutyrate (BHB) inhibits mineralisation, while acetoacetate activates mineralisation. Bone resorption increases either via increased osteoclast activity, or higher levels of BHB which decreases the localised pH
[74]. This releases undercarboxylated osteocalcin (OCN), which would negatively regulate BHB synthesis through its action on regulating basal insulin secretion
[75][76]. This keeps ketones in the physiologically healthy range
[77]. The low glucose and insulin state that facilitates dynamic bone remodelling, produces opened-up spaces for osteoblasts to then begin matrix synthesis and renewed bone mineralisation activity. This is followed by transition into a new generation of fresh osteocytes to maintain the newer sections of the lacuna-canalicular network. However, if a glucose restriction phase does not occur, over time the pre-osteoblast population may become compromised, whilst over-fossilisation simultaneously occurs. Subsequent chronic hyperinsulinaemia induces excess osteocytic ROS production to a degree that osteocytes are no longer able to actively inhibit mineralisation of their lacunocanalicular space, which is required to enable their survival, as well as those they are further connected with. Combined, these effects may drive increasing bone density, while making the bones both brittle and fragile.
9. Pyrophosphate and Sclerostin
Osteocytes produce pyrophosphate (ePPi), an inhibitor of mineralisation and carbonate solubiliser of bone mineral and matrix, thus regulating mineralisation and the circulatory systemic levels of calcium and phosphate
[78]. Hyperinsulinaemia increases mtROS and cellular pathology, compromising osteocyte health, leading to mitochondrial mediated living fossilisation of osteocytes within their lacunae. Magnesium-dependent spherite-mediated osteocyte mineralisation provides evidence to support this, where higher levels of magnesium are found in these micropetrosis fossilisation spaces formerly inhabited by osteocytes
[20]. This potentially traps/entombs magnesium within the living fossilised bone; subsequently contributing to magnesium deficiency (MgD). Furthermore, MgD has its own effects on potentiating hyperinsulinaemia
[79][80][81]. Culminating in a vicious feedforward cycle.
While bisphosphonates are known to work at inhibiting osteoclast bone resorption, they likely also work as an ePPi analogue. By assisting in inhibiting excessive mineralisation of the osteocytes lacunae, helping to maintain fluid flow and osteocyte connectivity where struggling osteocytes may fail to do so, thus saving the day in “assisting” the osteocytes in their job to be able to regulate dynamic bone turnover, a marker of metabolic health
[39][82].
A density threshold level of osteocytes is required to maintain homeostatic bone remodelling, from sensing damage in-order to initiate repair processes
[42], to mineral storage during pregnancy or in preparation for a winter with decreased food availability. Greater lacunae mineralisation has been detected with advanced age and/or untreated osteoporosis. Increased lacunae calcium content coupled with poorer quality matrix, contributes to increased bone brittleness and subsequent fragility
[19][40].
Osteocytes synthesise sclerostin, a 22.5 kDa protein, which reduces osteoblast differentiation, consequently downregulating bone metabolism. Although there is a clear relationship between hyperinsulinaemia and sclerostin levels, further research is required. Theoretically, a decreased osteocyte mass should predict lower sclerostin levels. However, elevated sclerostin levels have been found in patients with CVD and chronic kidney disease (CKD), two groups who typically have osteofragilitas. Furthermore, osteogenic differentiation of vascular smooth muscle cells (VSMC) in CVD and CKD patients have been detected, where there is an increase in expression of sclerostin in aortic valve tissue
[83][84][85][86]. This means caution is required when assessing bone and metabolic health via sclerostin levels.
10. Osteocytes Produce Alkaline Phosphatase
Alkaline phosphatase (ALP) catalyses the hydrolysis of inorganic pyrophosphate (PPi) to phosphate (Pi). Phosphate forms part of calcium hydroxyapatite crystals, and an increase in phosphate promotes mineralisation [87]. Nucleotide pyrophosphatase phosphodiesterase (NPP1) inhibits the action of ALP by increasing the concentration of calcification inhibitor pyrophosphate (ePPi) [69]. Insulin and the fed-state reduces NPP1 gene (Enpp1) expression. Fasting increases the concentration of ePPi via NPP1, leading to inhibition of excessive mineralisation of the osteocyte lacunocanalicular space, which results in maintaining osteocyte viability, dendritic connectivity and consequent dynamic bone remodelling capacity. Insulin action leads to decreased ePPi concentration, subsequently decreasing the osteocytes ability to inhibit bone mineralisation, thus increased mineralisation of the osteocytes lacunocanalicular space occurs. The fasted state provokes the opposite effect, inhibition of mineralisation, and potentially enhances physiological levels of bone resorption via increased beta-hydroxybutyrate (BHB) [92].
If autumn were a time for humans to accumulate stored energy in preparation for a fasting period during winter, where foods available during autumn increase insulin secretion, activating energy storage mechanisms, logically mineral storage would also be required and may also be facilitated via insulin’s action on inhibiting NPP1 production of ePPi. This may provide an evolutionary explanation, where seasonal hyperinsulinaemia propagates increased osteocyte-lacunae mineralisation during autumn, as an adaptive survival mechanism. This would then be followed by a winter of fasting, which would subsequently lead to increased NPP1 activity, relinquishing back into the system the stored minerals. Hyperinsulinaemia T2DM could be described as a metabolic phenotype reflecting a constantly “fed-state”, the ever-lasting autumn. Thus, providing an explanation of the normal to increased BMD observed in people with T2DM.
11. Osteocalcin
Patients with T2DM and insulin resistance have significantly lower levels of circulating osteocalcin (OCN) than healthy controls
[67][88][89][90][91]. OCN is a non-collagenous protein, largely synthesized by osteoblasts and osteocytes that retain their expression of OCN
[92]. Levels serve as a marker of osteoblast and osteocyte health. Serum OCN levels positively correlates with: dynamic bone remodelling, decreased insulin resistance (IR), and reduced T2DM and CVD risk
[91][93][94][95]. Much published research show OCN increases insulin secretion, with conclusions stating decreased OCN levels would result in decreased insulin synthesis and secretion, and result in impaired glucose homeostasis. However, when assessed in humans, low levels of OCN tracks with hyperinsulinaemia
[91][96]. Ergo providing evidence that production of high levels of insulin does not require high levels of OCN.
12. Osteocalcin Endocrine Effects
Interestingly, OCN significantly increases insulin-independent glucose uptake, and even more so in the presence of insulin. This leads to increased insulin sensitivity, through reducing the amount of insulin required to facilitate glucose uptake
[93]. Additionally, OCN production increases the expression of mitochondrial uncoupling protein 1 (UCP1) in adipocytes, leading to increased thermogenesis and mitochondrial biogenesis, thus increasing glucose and fatty acid oxidation capacity. Furthermore, OCN increases adipocyte production of adiponectin. However, caution is required when interpreting experiments involving administering exogenous OCN in animal studies, in which these animals are fed diets that do not induce hyperinsulinaemia (obesogenic for that species), that would mimic the main human causal factor for T2DM. In the hyperinsulinaemic state, adiponectin receptors Adr1/2 are downregulated
[97]. The signalling dynamics and results elucidated from animal studies in which exogenous OCN is provided to metabolically normal, or genetically induced OCN deficient and/or OCN receptor KO mice, would likely not be the same as what would occur in hyperinsulinaemic humans with low OCN. For example, healthy insulin levels and insulin sensitivity plus exogenous OC
n = X, whilst hyperinsulinaemia and insulin resistance plus exogenous OC
n = Y.
When wild-type mice (healthy) were administered exogenous OCN, their adipose tissues were observed to have upregulated peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and adiponectin. Both adiponectin and PGC1α expression and activation, lead to increased beta-oxidation and correspond to improved metabolism, insulin sensitivity and glucose homeostasis
[98]. Effectively, metabolically healthy mice respond in a physiologically normal way to the OCN. The elevated OCN in this context, is thus able to further effect (positive feedforward) mechanisms that consolidate better glucose/fatty acid oxidation and ROS management. The marker then becomes a maker of good health. OCN levels and the carboxylated-to-undercarboxylated ratio (cOCN:ucOCN or Gla:Glu) act as surrogate markers of osteoblast and osteocyte health, and consequently bone quality, which are then able to actively function in endocrine homeostasis and metabolic health.
The effect of OCN on adipocytes include: improved insulin independent glucose uptake, increased adiponectin synthesis and “energy wastage” through thermogenesis, and decreased inflammatory cytokine production, leading to increased skeletal and muscular insulin sensitivity
[93]. Greater insulin sensitivity decreases the amount of insulin required to achieve glucose homeostasis. Individuals who maintain normo-insulin levels via restricting carbohydrate intake, would likely maintain healthier osteoblasts and osteocytes. This enables healthy endogenous OCN synthesis, as is seen in healthy controls. In the context of normo-insulin and insulin sensitivity plus OCN, the result is X. Which is OCN increasing adiponectin synthesis and signalling in a none hyperinsulinaemia and normoglycaemia contextual environment. Furthermore, a reduced requirement and secretion of insulin, decreases excessive mitochondrial ROS production and subsequent downstream cellular pathophysiological adaptations.
13. Carboxylation of Osteocalcin
Post translational modification alters OCN into one of 3 isoforms, which affects bioavailability and activity of the bone-derived hormone. γ-carboxylation of OCN occurs before secretion from osteoblasts and osteocytes. γ-glutamyl carboxylase (GGCX) enzyme activity is dependent on availability of its cofactor vitamin K (in the reduced state), in order to carboxylate OCN on glutamic acid residues: Glu17, 21 and 24. The carboxylated (cOCN or Gla-OCN) form, is the most abundant form in bone extracellular matrix as carboxylation increases OCN affinity for the mineral component of bone hydroxyapatite
[75][93]. The undercarboxylated and uncarboxylated forms, (ucOCN or Glu-OCN) are considered the biologically active hormone isoforms. All three isoforms are present in the blood
[57]. OCN concentration in human blood ranges between 10 to 40 ng/mL
[57]. Interestingly, in vitro experiments by Hill et al. show OCN in both carboxylated and uncarboxylated forms are biologically active, both are able to modulate glucose uptake and increase insulin sensitivity. However, the ucOCN form was more effective
[93].
14. Osteocalcin and Insulin
Much research has shown that insulin signalling induces osteoblasts and osteocytes to produce OCN and in vivo studies indicate that plasma OCN stimulates an increase in pancreatic beta cell differentiation/proliferation via increasing cyclin D1, D2 and Cdk4 gene expression, proteins involved in cell division
[98]. Additionally, OCN directly and indirectly via glucagon-like peptide-1 (GLP-1), increases insulin production capacity and secretion upon glucose stimulus
[98]. While this is shown in in vitro and animal studies, T1DM patients who are given exogenous insulin should technically gain in increased endogenous OCN synthesis, this would then be expected to stimulate pancreatic beta-cell proliferation and subsequent endogenous insulin secretion capabilities. However, T1DM patients do not appear to gain in the upregulation of endogenous insulin production. This may be due to a lack of pancreatic precursor beta-cells, although it has been shown that both T1DM and late-stage T2DM patients do have some functioning precursor pancreatic beta-cells
[95][99].
Wei et al. investigated the potential role of OCN as a means to stimulate pancreatic beta-cell proliferation, given T1DM patients retain a small residual population of functional beta-cells [99][100][101], the logic of their hypothesis seems plausible. T2DM patients have low OCN levels too. However, this is with high insulin levels in non-insulin dependent diabetes mellites (NIDDM). The question remains then, would these patients benefit from exogenous OCN therapy? Or similar to conditions of T2DM, patients given exogenous insulin, serves only to mask the downstream problem (hyperglycaemia), while increasing hyperinsulinaemia and insulin resistance, driving the disease further [88][102]. If OCN increases insulin secretion, then would we not see higher levels of OCN in hyperinsulinaemia pathologies such as T2DM, CVD and MetS? On the contrary, those with normal insulin levels have significantly higher OCN levels [88][102]. This may indicate OCN levels and the carboxylation ratio, are firstly markers and then contributory makers of bone fracture resistance, and metabolic and endocrine health.
Using a mouse model, Ferron et al. showed that intermittent injections of OCN improved glucose metabolism, and increased skeletal mitochondrial content which led to improved glucose and fatty acid oxidation capacity. Elevated OCN levels in the absence of hyperinsulinaemia also increased energy expenditure, corroborating other research showing OCN signalling via increasing adiponectin production, leads to increases in brown fat UCP1, resulting in increased thermogenesis. As fat and glucose is oxidised more efficiently, independent of insulin mediated glucose uptake, metabolic markers consequently improve
[103].
Evidence suggests the metabolic phenotype of low insulin and glucose levels facilitates maintaining healthy osteoblasts and osteocytes, dendritic connectivity and maintenance of the lacunae-canalicular network. This leads to the metabolic healthy phenotype of higher levels of OCN synthesis and carboxylation capacity, resulting in fracture-resistant bone, with osteocytes maintaining basal bone remodelling. Furthermore, the resultant ability to endogenously produce OCN, enables bone to participate in its endocrine-action on other tissues and organs, further contributing to improved glucose homeostasis and insulin sensitivity
[104]. Restriction of dietary carbohydrate intake simultaneously maintains low glucose levels and minimises additional exogenous stimulus on insulin secretion, thus maintaining both markers in the low healthy physiological range.
15. cOCN Levels Determine Hydroxyapatite Alignment Formation
Moriishi et al. in
PLOS Genetics, demonstrated using mouse model OCN knockouts of the two mouse genes for OCN:
Bglap and
Bglap2, unlike humans who poses only one OCN gene, that bone apatite crystallite alignment is dependent on carboxylated OCN
[105]. In the OCN-deficient mice, bone strength decreased, supporting the classical biology phrase—structure dictates function. In this case, bone strength is determined by its structural quality, which includes bone mass and quality of crystallite alignment with collagen fibres. These structural features, combined, determine bone fracture resistance or osteofragilitas
[106]. It is important to note that Moriishi was not investigating OCN deficiency in the context of hyperinsulinaemia, which likely would further contribute other detrimental factors, such as poorer collagen synthesis and increased collagen glycation damage.
Hyperinsulinaemia and hyperglycaemia propagation of impaired osteoblastogene-sis and osteocytogenesis results in decreased OCN production capacity, which impairs hydroxyapatite crystallographic orientation to collagen fibrils. Combined with poorer quality collagen synthesis and increased glycated collagen, the summative results may lead to compounding effects on bone fragility via compromised structural quality, that is independent of BMD. The sum of all of these dysregulated/impaired conditions, likely leads to the increased fracture rates seen in patients with the T2DM hyperinsulinaemia- osteofragilitas phenotype.
16. Osteocalcin Regulation of Ketosis
Fasting increases ketogenesis and plasma BHB which increases bone resorption ability via lowering the localised pH level as well as inhibiting/regulating osteoblast mineralisation activity
[74]. This would increase plasma OCN levels that would go on to stimulate pancreatic beta cell proliferation and insulin production capacity. Natural diurnal cortisol signalling leads to hepatic glycogenolysis and release of glucose to the system, in turn stimulating insulin secretion. Together, through BHB effect on OCN release, they provide the stimulus for insulin secretion to act as a feedback mechanism to regulate ketogenesis.
OCN is considered to protect against obesity, improve glucose uptake and enhance insulin sensitivity, either directly or via OCN stimulated adiponectin secretion
[92][95][98][107][108][109]. Osteocytes comprise the largest population of bone cells, making them likely the biggest producer of OCN than is currently understood. Logically, a substantial loss of osteocytes would result in decreased OCN production and lead to subsequent increases in adiposity. This phenotype is seen in HI/T2DM/CVD patients, who have lower plasma OCN and adiponectin, with increased or normal BMD. Bone mineralisation is possible without OCN and may be enhanced in the hyperinsulinaemic state
[105]. Micropetrosis/living-fossilisation of the osteocytic lacuna-canalicular has been demonstrated to be increased in hyperinsulinaemia pathologies
[19][20][22][25]. BHB mediated inhibition of mineralisation may be analogous to ePPi and bisphosphonates that help osteocytes maintain their lacunocanalicular network. Inhibition of lacunocanalicular mineralisation concurrent with maintaining osteocyte viability, results in correctly-formed fracture resistant bone that is also dynamically remodelled, a function of healthy bone metabolism leading to wider effects on whole body metabolism.
17. Vitamin D and Magnesium
Vitamin D is required for osteocyte viability and dendrite connectivity [110]. Hyperinsulinaemia reduces vitamin D availability by increasing sequestration of the lipophilic hormone into adipocytes [111]. Patients with pathologies of hyperinsulinaemia, including T2DM, CVD, obesity, MetS and metabolic cancers, are associated with having a lower vitamin D status [90][112]. In addition, hyperinsulinaemia promotes magnesium deficiency (MgD) which decreases vitamin D transport in the blood [80]. Chronic hyperinsulinaemia dysregulated vitamin D metabolism negatively affects osteocyte health and their subsequent ability to perform dynamic perilacunar remodelling [110].
Insulin is required for healthy bone mineralisation, as seen in insulin insufficient T1DM [113]. Puberty and pregnancy are two stages of development where a natural state of hyperinsulinaemia occurs [73][114][115]. Hyperinsulinaemia enables increased growth in bones during puberty, and mineral accretion during pregnancy, which may be in the form of micropetrosis (osteocyte-lacunae mineralisation), to ensure adequate provision of minerals for nursing offspring during lactation. These phases of life lend a physiological adaptive explanation as to why we would see an increase in BMD in T2DM hyperinsulinaemia [27][29][116]. However, puberty and pregnancy are for a limited duration of time and come with either growth in bones (puberty), or lactation and subsequent resorption of bone minerals. Whereas T2DM hyperinsulinaemia may go undetected for many years, a pernicious chronic elevation, leading to excessive micropetrosis and living fossilisation. The resultant disconnecting of osteocytes from one another, impairs their ability to: sense and transmit information, modulate one another, and directly/indirectly modulate bone turnover [28][58][117]. This hypothesis provides a plausible explanation as to why we would see normal to H-BMD in T2DM not conferring fracture resistance.
The article is from 10.3390/biomedicines9091165